


Patient Information ⮝
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use). Review the steps for direct patient administration with patients and caregivers. Training by the healthcare provider should aim to ensure that patients and caregivers can successfully perform all of the steps in the Instructions for Use of NEUPOGEN vial and prefilled syringe, including showing the patient or caregiver how to measure the required dose, particularly if a patient is on a dose other than the entire prefilled syringe. If a patient or caregiver is not able to demonstrate that they can measure the dose and administer the product successfully, you should consider whether the patient is an appropriate candidate for self-administration of NEUPOGEN or whether the patient would benefit from a different NEUPOGEN presentation.
Advise Patients Of The Following Risks And Potential Risks With Neupogen: ⮝
- Rupture or enlargement of the spleen may occur. Symptoms include left upper quadrant abdominal pain or left shoulder pain. Advise patients to report pain in these areas to their physician immediately[see Warnings and Precautions (5.1)].
- Dyspnea, with or without fever, progressing to Acute Respiratory Distress Syndrome, may occur. Advise patients to report dyspnea to their physician immediately[see Warnings and Precautions (5.2)].
- Serious allergic reactions may occur, which may be signaled by rash facial edema wheezing dyspnea hypotension or tachycardia. Advise patients to seek immediate medical attention if signs or symptoms of hypersensitivity reaction occur[see Warnings and Precautions (5.3)].
- In patients with sickle cell disease, sickle cell crisis and death have occurred. Discuss potential risks and benefits for patients with sickle cell disease prior to the administration of human granulocyte colony-stimulating factors[see Warnings and Precautions (5.4)].
- Glomerulonephritis may occur. Symptoms include swelling of the face or ankles, dark colored urine or blood in the urine, or a decrease in urine production. Advise patients to report signs or symptoms of glomerulonephritis to their physician immediately[see Warnings and Precautions (5.5)].
- Cutaneous vasculitis may occur, which may be signaled by purpura or erythema. Advise patients to report signs or symptoms of vasculitis to their physician immediately[see Warnings and Precautions (5.11)].
- Aortitis may occur. Symptoms may include fever, abdominal pain, malaise, back pain, and increased inflammatory markers. Advise patients to report signs and symptoms of aortitis to their physician immediately[see Warnings and Precautions (5.15)].
Advise patients acutely exposed to myelosuppressive doses of radiation (Hematopoietic Syndrome of Acute Radiation Syndrome) that efficacy studies of NEUPOGEN for this indication could not be conducted in humans for ethical and feasibility reasons and that, therefore, approval of this use was based on efficacy studies conducted in animals[see Clinical Studies (14.6)].
Instruct Patients Who Self-administer Neupogen Using The Prefilled Syringe Or Single-dose Vial Of The: ⮝
- Importance of following the applicable Instructions for Use.
- Dangers of reusing needles, syringes, or unused portions of single-dose vials.
- Importance of following local requirements for proper disposal of used syringes, needles, and unused vials.
- Importance of informing the healthcare provider if difficulty occurs when measuring or administering partial contents of the NEUPOGEN prefilled syringe. If difficulty occurs, use of the NEUPOGEN vial may be considered.
- Difference in product concentration of the NEUPOGEN prefilled syringe in comparison to the NEUPOGEN vial. When switching patients from the NEUPOGEN prefilled syringe to the NEUPOGEN vial, or vice versa, ensure that patients understand the correct volume to be administered since the concentration of NEUPOGEN differs between the prefilled syringe and the vial.
NEUPOGEN(filgrastim)
Manufactured By: ⮝
Amgen Inc.
One Amgen Center Drive
Thousand Oaks, California 91320-1799
U.S. License Number 1080Patent:http://pat.amgen.com/neupogen/
1991-2018 Amgen Inc. All rights reserved.
www.NEUPOGEN.com
1-800-77-AMGEN (1-800-772-6436)1xxxxxx
v31
- No Title 1572554794
- Highlights Of Prescribing Information
- Recent Major Changes
- Indications And Usage
- Dosage And Administration
- Dosage Forms And Strengths
- Contraindications
- Warnings And Precautions
- Adverse Reactions
- 1 Indications And Usage
- 2 Dosage And Administration
- 5 Warnings And Precautions
- 6 Adverse Reactions
- 8 Use In Specific Populations
- 12 Clinical Pharmacology
- 13 Nonclinical Toxicology
- 14 Clinical Studies
- 1 Indications And Usage
- 2 Dosage And Administration
- 3 Dosage Forms And Strengths
- 4 Contraindications
- 5 Warnings And Precautions
- 6 Adverse Reactions
- 8 Use In Specific Populations
- 10 Overdosage
- 11 Description
- 12 Clinical Pharmacology
- 13 Nonclinical Toxicology
- 14 Clinical Studies
- 16 How Supplied/storage And Handling
- No Title 1572449134
- Principal Display Panel
- Description
- Clinical Pharmacology
- Clinical Experience
- Warnings
- Precautions
- Overdosage
- How Supplied
- References
No Title 1572554794 ⮝
Instructions for Use
NEUPOGEN (nu-po-jen)
(filgrastim)
Injection
Single-Dose VialImportant
Read the Patient Information for important information you need to know about NEUPOGEN before using these Instructions for Use.
Before you use a NEUPOGEN vial, read this important information:
Storing your NEUPOGEN vial
- Store the vial in the refrigerator between 36 F to 46 F (2 C to 8 C).
- Do not freeze.
- Keep the vial in the original carton to protect from light or physical damage.
- Take the vial out of the refrigerator 30 minutes before use and allow it to reach room temperature before preparing an injection.
- Throw away (dispose of) any vial that has been left at room temperature for longer than 24 hours.
- After you inject your dose, throw away (dispose of) any unused NEUPOGEN left in the vial. Do not save unused NEUPOGEN in the vial for later use.
- Keep the NEUPOGEN vial out of the reach of children.
Using your vial
- It is important that you do not try to give the injection unless you or your caregiver has received training from your healthcare provider.
- Make sure the name NEUPOGEN appears on the carton and vial label.
- Only use the vial 1 time. Discard (throw away) the vial with any remaining NEUPOGEN liquid.
- Do not use a vial after the expiration date on the label.
- Do not shake the vial.
- Do not use the vial if the medicine is cloudy or discolored or contains flakes or particles.
Call your healthcare provider if you have any questions.
Step 1: Prepare
A Remove the vial from the refrigerator.
On a clean, well-lit surface, place the vial at room temperature for 30 minutes before you give an injection.
- Do not try to warm the vial by using a heat source such as hot water or microwave.
- Do not leave the vial in direct sunlight.
- Do not shake the vial.
- Use the vial only 1 time.
B Inspect the vial.
Make sure the medicine in the vial is clear and colorless.
- Do not use the vial if:
The medicine is cloudy or discolored or contains flakes or particles.
The expiration date printed on the label has passed.
- In all cases, use a new vial and call your healthcare provider.
C Gather all materials needed for your injection.
Wash your hands thoroughly with soap and water.
On a clean, well-lit work surface, place the:
- Vial
- Disposable syringe and needle
- 2 alcohol wipes
- Cotton ball or gauze pad
- Adhesive bandage
Sharps disposal container
- Only use the disposable syringes and needles that your healthcare provider prescribes.
- Only use the syringes and needles 1 time. Discard (throw away) any used syringes and needles.
- You should only use a syringe that is marked in tenths of milliliters (mL).
- Your healthcare provider will show you how to measure the correct dose of NEUPOGEN. This dose will be measured in milliliters (mL).
Step 2: Get Ready
D Take the cap off the vial. Clean the rubber stopper with one alcohol wipe.

E Check the carton containing the syringe. If the carton has been opened or damaged, do not use that syringe. Dispose of (throw away) that syringe in the sharps disposal container.
F Hold the syringe by the barrel with the needle cap pointing up. Carefully pull the needle cap straight off and away from your body.

Pull back on the plunger and draw air into the syringe that is the same amount (mL) as the dose of NEUPOGEN that your healthcare provider prescribed.
Important: Throw the needle cap into the sharps disposal container.
G Keep the vial on the flat working surface and insert the needle straight down through the rubber stopper. Do not insert the needle through the rubber stopper more than 1 time.
H Push the plunger down and inject all the air from the syringe into the vial of NEUPOGEN.
I Keep the needle in the vial and turn the vial upside down. Make sure that the NEUPOGEN liquid is covering the tip of the needle.
J Keep the vial upside down and slowly pull back on the plunger to fill the syringe barrel with NEUPOGEN to the correct marking amount (mL) of medicine that matches the dose your healthcare provider prescribed.
K Keep the needle in the vial and check for air bubbles in the syringe. If there are air bubbles, gently tap the syringe barrel with your finger until the air bubbles rise to the top. Slowly push the plunger up to push the air bubbles out of the syringe.
L Keep the tip of the needle in the liquid and again pull the plunger back to the number on the syringe barrel that matches your dose. Check again for air bubbles. The air in the syringe will not hurt you, but too large an air bubble can reduce your dose of NEUPOGEN. If there are still air bubbles, repeat the steps above to remove them.
M Check again to make sure that you have the correct dose in the syringe. It is important that you use the exact dose prescribed by your healthcare provider. Do not remove the needle from the vial. Lay the vial down on its side with the needle still in the vial.
Step 3: Select and Prepare the Injection Site
N Prepare and clean your injection site.
You can use:
- Thigh
- Stomach area (abdomen), except for a 2-inch area right around your navel (belly button)
- Upper outer area of your buttocks (only if someone else is giving you the injection)
- Outer area of upper arm (only if someone else is giving you the injection)
Clean your injection site with a clean alcohol wipe.
- Let your skin dry.
- Do not touch this area again before injecting.
- If you want to use the same injection site, make sure it is not the same spot on the injection site area you used for a previous injection.
- Do not inject into areas where the skin is tender, bruised, red, or hard. Avoid injecting into areas with scars or stretch marks.
Step 4: Subcutaneous (under the skin) injection
O Remove the prepared syringe and needle from the vial.
P Pinch your injection site to create a firm surface.
Important: Keep skin pinched while injecting.
Q Hold the pinch. Insert the needle into the skin at a 45 to 90 degree angle.
R Using slow and constant pressure, push the plunger until it reaches the bottom.
When done gently pull the syringe off of your skin.
Step 5: Finish
S Discard (throw away) the used syringe and vial.
- Put your used syringes, needles, and vials in a FDA-cleared sharps disposal container right away after use. Do not throw away (dispose of) needles, syringes and vials in your household trash.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that:
- is made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- is upright and stable during use,
- is leak-resistant, and
- is properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used syringes and needles. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA s website at: http://www.fda.gov/safesharpsdisposal.
- Do not reuse the syringe or vial.
- Do not recycle the syringe, vial, or sharps disposal container or throw them into household trash.
Important: Always keep the sharps disposal container out of the reach of children.
T Examine the injection site.
If there is blood, press a cotton ball or gauze pad on your injection site. Do not rub the injection site. Apply an adhesive bandage if needed.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
NEUPOGEN (filgrastim)
Manufactured by:Amgen Inc.
One Amgen Center Drive
Thousand Oaks, California 91320-1799 U.S.A.U.S. License No. 1080
Revised: 06/2016
1xxxxxx
1991-2016 Amgen Inc. All rights reserved.
v1
Highlights Of Prescribing Information ⮝
These highlights do not include all the information needed to use NEUPOGEN safely and effectively. See full prescribing information for NEUPOGEN.
NEUPOGEN (filgrastim) injection, for subcutaneous or intravenous use
Initial U.S. Approval: 1991
Recent Major Changes ⮝
Warnings and Precautions: Aortitis (5.15) 06/2018
Indications And Usage ⮝
NEUPOGEN is a leukocyte growth factor indicated to
- Decrease the incidence of infection as manifested by febrile neutropenia in patients with nonmyeloid malignancies receiving myelosuppressive anti-cancer drugs associated with a significant incidence of severe neutropenia with fever (1.1)
- Reduce the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy treatment of patients with acute myeloid leukemia (AML) (1.2)
- Reduce the duration of neutropenia and neutropenia-related clinical sequelae e.g. febrile neutropenia, in patients with nonmyeloid malignancies undergoing myeloablative chemotherapy followed by bone marrow transplantation (BMT) (1.3)
- Mobilize autologous hematopoietic progenitor cells into the peripheral blood for collection by leukapheresis (1.4)
- Reduce the incidence and duration of sequelae of severe neutropenia (e.g. fever infections oropharyngeal ulcers) in symptomatic patients with congenital neutropenia cyclic neutropenia or idiopathic neutropenia (1.5)
- Increase survival in patients acutely exposed to myelosuppressive doses of radiation (Hematopoietic Syndrome of Acute Radiation Syndrome) (1.6)
Dosage And Administration ⮝
- Patients with cancer receiving myelosuppressive chemotherapy or induction and/or consolidation chemotherapy for AML
Recommended starting dose is 5 mcg/kg/day subcutaneous injection, short intravenous infusion (15 to 30 minutes), or continuous intravenous infusion. See Full Prescribing Information for recommended dosage adjustments and timing of administration (2.1)
- Patients with cancer undergoing bone marrow transplantation
10 mcg/kg/day given as an intravenous infusion no longer than 24 hours. See Full Prescribing Information for recommended dosage adjustments and timing of administration (2.2)
- Patients undergoing autologous peripheral blood progenitor cell collection and therapy
10 mcg/kg/day subcutaneous injection (2.3)
Administer for at least 4 days before first leukapheresis procedure and continue until last leukapheresis (2.3)
- Patients with congenital neutropenia
Recommended starting dose is 6 mcg/kg subcutaneous injection twice daily (2.4)
- Patients with cyclic or idiopathic neutropenia
Recommended starting dose is 5 mcg/kg subcutaneous injection daily (2.4)
- Patients acutely exposed to myelosuppressive doses of radiation
10 mcg/kg/day subcutaneous injection (2.5)
Dosage Forms And Strengths ⮝
Vial
Prefilled Syringe
Contraindications ⮝
- Patients with a history of serious allergic reactions to human granulocyte colony-stimulating factors such as filgrastim or pegfilgrastim. (4)
Warnings And Precautions ⮝
- Fatal splenic rupture: Evaluate patients who report left upper abdominal or shoulder pain for an enlarged spleen or splenic rupture. (5.1)
- Acute respiratory distress syndrome (ARDS): Evaluate patients who develop fever and lung infiltrates or respiratory distress for ARDS. Discontinue NEUPOGEN in patients with ARDS. (5.2)
- Serious allergic reactions, including anaphylaxis: Permanently discontinue NEUPOGEN in patients with serious allergic reactions. (5.3)
- Fatal sickle cell crises: Have occurred. (5.4)
- Glomerulonephritis: Evaluate and consider dose-reduction or interruption of NEUPOGEN if causality is likely. (5.5)
Adverse Reactions ⮝
Most common adverse reactions in patients:
- With nonmyeloid malignancies receiving myelosuppressive anti-cancer drugs ( 5% difference in incidence compared to placebo) are pyrexia, pain, rash, cough, and dyspnea. (6.1)
- With AML ( 2% difference in incidence) are pain, epistaxis and rash. (6.1)
- With nonmyeloid malignancies undergoing myeloablative chemotherapy followed by BMT ( 5% difference in incidence) is rash. (6.1)
- Undergoing peripheral blood progenitor cell mobilization and collection ( 5% incidence) are bone pain, pyrexia and headache. (6.1)
- With severe chronic neutropenia (SCN) ( 5% difference in incidence) are pain, anemia, epistaxis, diarrhea, hypoesthesia and alopecia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Amgen Medical Information at 1-800-77-AMGEN (1-800-772-6436) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 6/2018
1 Indications And Usage ⮝
1.1 Patients with Cancer Receiving Myelosuppressive Chemotherapy
1.2 Patients with Acute Myeloid Leukemia Receiving Induction or Consolidation Chemotherapy
1.3 Patients with Cancer Undergoing Bone Marrow Transplantation
1.4 Patients Undergoing Autologous Peripheral Blood Progenitor Cell Collection and Therapy
1.5 Patients with Severe Chronic Neutropenia
1.6 Patients Acutely Exposed to Myelosuppressive Doses of Radiation (Hematopoietic Syndrome of Acute Radiation Syndrome)
2 Dosage And Administration ⮝
2.1 Dosage in Patients with Cancer Receiving Myelosuppressive Chemotherapy or Induction and/or Consolidation Chemotherapy for AML
2.2 Dosage in Patients with Cancer Undergoing Bone Marrow Transplantation
2.3 Dosage in Patients Undergoing Autologous Peripheral Blood Progenitor Cell Collection and Therapy
2.4 Dosage in Patients with Severe Chronic Neutropenia
2.5 Dosage in Patients Acutely Exposed to Myelosuppressive Doses of Radiation (Hematopoietic Syndrome of Acute Radiation Syndrome)
2.6 Important Administration Instructions
5 Warnings And Precautions ⮝
5.1 Splenic Rupture
5.2 Acute Respiratory Distress Syndrome
5.3 Serious Allergic Reactions
5.4 Sickle Cell Disorders
5.5 Glomerulonephritis
5.6 Alveolar Hemorrhage and Hemoptysis
5.7 Capillary Leak Syndrome
5.8 Patients with Severe Chronic Neutropenia
5.9 Thrombocytopenia
5.10 Leukocytosis
5.11 Cutaneous Vasculitis
5.12 Potential Effect on Malignant Cells
5.13 Simultaneous Use with Chemotherapy and Radiation Therapy Not Recommended
5.14 Nuclear Imaging
5.15 Aortitis
6 Adverse Reactions ⮝
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
8 Use In Specific Populations ⮝
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
12 Clinical Pharmacology ⮝
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 Nonclinical Toxicology ⮝
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and Pharmacology
14 Clinical Studies ⮝
14.1 Patients with Cancer Receiving Myelosuppressive Chemotherapy
14.2 Patients with Acute Myeloid Leukemia Receiving Induction or Consolidation Chemotherapy
14.3 Patients with Cancer Undergoing Bone Marrow Transplantation
14.4 Patients Undergoing Autologous Peripheral Blood Progenitor Cell Collection and Therapy
14.5 Patients with Severe Chronic Neutropenia
14.6 Patients Acutely Exposed to Myelosuppressive Doses of Radiation (Hematopoietic Syndrome of Acute Radiation Syndrome)
1 Indications And Usage ⮝
1.1 Patients with Cancer Receiving Myelosuppressive Chemotherapy
NEUPOGEN is indicated to decrease the incidence of infection as manifested by febrile neutropenia in patients with nonmyeloid malignancies receiving myelosuppressive anti-cancer drugs associated with a significant incidence of severe neutropenia with fever [see Clinical Studies (14.1)].
1.2 Patients with Acute Myeloid Leukemia Receiving Induction or Consolidation Chemotherapy
NEUPOGEN is indicated for reducing the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy treatment of patients with acute myeloid leukemia (AML) [see Clinical Studies (14.2)].
1.3 Patients with Cancer Undergoing Bone Marrow Transplantation
NEUPOGEN is indicated to reduce the duration of neutropenia and neutropenia-related clinical sequelae e.g. febrile neutropenia, in patients with nonmyeloid malignancies undergoing myeloablative chemotherapy followed by bone marrow transplantation [see Clinical Studies (14.3)].
1.4 Patients Undergoing Autologous Peripheral Blood Progenitor Cell Collection and Therapy
NEUPOGEN is indicated for the mobilization of autologous hematopoietic progenitor cells into the peripheral blood for collection by leukapheresis [see Clinical Studies (14.4)].
1.5 Patients with Severe Chronic Neutropenia
NEUPOGEN is indicated for chronic administration to reduce the incidence and duration of sequelae of neutropenia (e.g. fever infections oropharyngeal ulcers) in symptomatic patients with congenital neutropenia cyclic neutropenia or idiopathic neutropenia [see Clinical Studies (14.5)].
1.6 Patients Acutely Exposed to Myelosuppressive Doses of Radiation (Hematopoietic Syndrome of Acute Radiation Syndrome)
NEUPOGEN is indicated to increase survival in patients acutely exposed to myelosuppressive doses of radiation [see Clinical Studies (14.6)].
2 Dosage And Administration ⮝
2.1 Dosage in Patients with Cancer Receiving Myelosuppressive Chemotherapy or Induction and/or Consolidation Chemotherapy for AML
The recommended starting dosage of NEUPOGEN is 5 mcg/kg/day administered as a single daily injection by subcutaneous injection by short intravenous infusion (15 to 30 minutes) or by continuous intravenous infusion. Obtain a complete blood count (CBC) and platelet count before instituting NEUPOGEN therapy and monitor twice weekly during therapy. Consider dose escalation in increments of 5 mcg/kg for each chemotherapy cycle according to the duration and severity of the absolute neutrophil count (ANC) nadir. Recommend stopping NEUPOGEN if the ANC increases beyond 10 000/mm3 [see Warnings and Precautions (5.10)].
Administer NEUPOGEN at least 24 hours after cytotoxic chemotherapy. Do not administer NEUPOGEN within the 24-hour period prior to chemotherapy [see Warnings and Precautions (5.13)]. A transient increase in neutrophil count is typically seen 1 to 2 days after initiation of NEUPOGEN therapy. Therefore, to ensure a sustained therapeutic response administer NEUPOGEN daily for up to 2 weeks or until the ANC has reached 10 000/mm3 following the expected chemotherapy-induced neutrophil nadir. The duration of NEUPOGEN therapy needed to attenuate chemotherapy-induced neutropenia may be dependent on the myelosuppressive potential of the chemotherapy regimen employed.
2.2 Dosage in Patients with Cancer Undergoing Bone Marrow Transplantation
The recommended dosage of NEUPOGEN following bone marrow transplantation (BMT) is 10 mcg/kg/day given as an intravenous infusion no longer than 24 hours. Administer the first dose of NEUPOGEN at least 24 hours after cytotoxic chemotherapy and at least 24 hours after bone marrow infusion. Monitor CBCs and platelet counts frequently following marrow transplantation.
During the period of neutrophil recovery titrate the daily dosage of NEUPOGEN against the neutrophil response (see Table 1).
Table 1. Recommended Dosage Adjustments During Neutrophil Recovery in Patients with Cancer Following BMT Absolute Neutrophil Count NEUPOGEN Dosage Adjustment When ANC greater than 1,000/mm3 for 3
consecutive daysReduce to 5 mcg/kg/daya Then, if ANC remains greater than 1,000/mm3 for
3 more consecutive daysDiscontinue NEUPOGEN Then, if ANC decreases to less than 1,000/mm3 Resume at 5 mcg/kg/day a If ANC decreases to less than 1,000/mm3 at any time during the 5 mcg/kg/day administration increase NEUPOGEN to 10 mcg/kg/day and then follow the above steps.
2.3 Dosage in Patients Undergoing Autologous Peripheral Blood Progenitor Cell Collection and Therapy
The recommended dosage of NEUPOGEN for the mobilization of autologous peripheral blood progenitor cells (PBPC) is 10 mcg/kg/day given by subcutaneous injection. Administer NEUPOGEN for at least 4 days before the first leukapheresis procedure and continue until the last leukapheresis. Although the optimal duration of NEUPOGEN administration and leukapheresis schedule have not been established administration of NEUPOGEN for 6 to 7 days with leukaphereses on days 5 6 and 7 was found to be safe and effective [see Clinical Studies (14.4)]. Monitor neutrophil counts after 4 days of NEUPOGEN and discontinue NEUPOGEN if the white blood cell (WBC) count rises to greater than 100 000/mm3.
2.4 Dosage in Patients with Severe Chronic Neutropenia
Prior to starting NEUPOGEN in patients with suspected chronic neutropenia, confirm the diagnosis of severe chronic neutropenia (SCN) by evaluating serial CBCs with differential and platelet counts and evaluating bone marrow morphology and karyotype. The use of NEUPOGEN prior to confirmation of a correct diagnosis of SCN may impair diagnostic efforts and may thus impair or delay evaluation and treatment of an underlying condition other than SCN causing the neutropenia.
The recommended starting dosage in patients with Congenital Neutropenia is 6 mcg/kg as a twice daily subcutaneous injection and the recommended starting dosage in patients with Idiopathic or Cyclic Neutropenia is 5 mcg/kg as a single daily subcutaneous injection.
Dosage Adjustments in Patients with Severe Chronic Neutropenia
Chronic daily administration is required to maintain clinical benefit. Individualize the dosage based on the patient s clinical course as well as ANC. In the SCN postmarketing surveillance study, the reported median daily doses of NEUPOGEN were: 6 mcg/kg (congenital neutropenia), 2.1 mcg/kg (cyclic neutropenia), and 1.2 mcg/kg (idiopathic neutropenia). In rare instances, patients with congenital neutropenia have required doses of NEUPOGEN greater than or equal to 100 mcg/kg/day.
Monitor CBCs for Dosage Adjustments
During the initial 4 weeks of NEUPOGEN therapy and during the 2 weeks following any dosage adjustment monitor CBCs with differential and platelet counts. Once a patient is clinically stable monitor CBCs with differential and platelet counts monthly during the first year of treatment. Thereafter, if the patient is clinically stable, less frequent routine monitoring is recommended.
2.5 Dosage in Patients Acutely Exposed to Myelosuppressive Doses of Radiation (Hematopoietic Syndrome of Acute Radiation Syndrome)
The recommended dose of NEUPOGEN is 10 mcg/kg as a single daily subcutaneous injection for patients exposed to myelosuppressive doses of radiation. Administer NEUPOGEN as soon as possible after suspected or confirmed exposure to radiation doses greater than 2 gray (Gy).
Estimate a patient s absorbed radiation dose (i.e., level of radiation exposure) based on information from public health authorities, biodosimetry if available, or clinical findings such as time to onset of vomiting or lymphocyte depletion kinetics.
Obtain a baseline CBC and then serial CBCs approximately every third day until the ANC remains greater than 1,000/mm3 for 3 consecutive CBCs. Do not delay administration of NEUPOGEN if a CBC is not readily available.
Continue administration of NEUPOGEN until the ANC remains greater than 1,000/mm3 for 3 consecutive CBCs or exceeds 10,000/mm3 after a radiation-induced nadir.
2.6 Important Administration Instructions
NEUPOGEN is supplied in single-dose vials (for subcutaneous use or intravenous infusion) and single-dose prefilled syringes (for subcutaneous use) [see Dosage Forms and Strengths (3)]. Prior to use remove the vial or prefilled syringe from the refrigerator and allow NEUPOGEN to reach room temperature for a minimum of 30 minutes and a maximum of 24 hours. Discard any vial or prefilled syringe left at room temperature for greater than 24 hours. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit (the solution is clear and colorless). Do not administer NEUPOGEN if particulates or discoloration are observed.
Discard unused portion of NEUPOGEN in vials or prefilled syringes; do not re-enter the vial. Do not save unused drug for later administration.
Subcutaneous Injection
Inject NEUPOGEN subcutaneously in the outer area of upper arms, abdomen, thighs, or upper outer areas of the buttock. If patients or caregivers are to administer NEUPOGEN, instruct them in appropriate injection technique and ask them to follow the subcutaneous injection procedures in the Instructions for Use for the vial or prefilled syringe [see Patient Counseling Information (17)].
Training by the healthcare provider should aim to demonstrate to those patients and caregivers how to measure the dose of NEUPOGEN, and the focus should be on ensuring that a patient or caregiver can successfully perform all the steps in the Instructions for Use for the vial or prefilled syringe. If a patient or caregiver is not able to demonstrate that they can measure the dose and administer the product successfully, you should consider whether the patient is an appropriate candidate for self-administration of NEUPOGEN or whether the patient would benefit from a different NEUPOGEN presentation. If a patient or caregiver experiences difficulty measuring the required dose, especially if it is other than the entire contents of the NEUPOGEN prefilled syringe, use of the NEUPOGEN vial may be considered.
If the patient or caregiver misses a dose of NEUPOGEN, instruct them to contact their healthcare provider.
Administration Instructions for the Prefilled Syringe
Persons with latex allergies should not administer the NEUPOGEN prefilled syringe, because the needle cap contains dry natural rubber (derived from latex).
Administration Instructions for Dilution (Vial Only)
If required for intravenous administration NEUPOGEN (vial only) may be diluted in 5% Dextrose Injection, USP from a concentration of 300 mcg/mL to 5 mcg/mL (do not dilute to a final concentration less than 5 mcg/mL). NEUPOGEN diluted to concentrations from 5 mcg/mL to 15 mcg/mL should be protected from adsorption to plastic materials by the addition of Albumin (Human) to a final concentration of 2 mg/mL. When diluted in 5% Dextrose Injection, USP or 5% Dextrose plus Albumin (Human) NEUPOGEN is compatible with glass bottles polyvinyl chloride (PVC) and polyolefin intravenous bags and polypropylene syringes. Do not dilute with saline at any time because the product may precipitate.
Diluted NEUPOGEN solution can be stored at room temperature for up to 24 hours. This 24-hour time period includes the time during room temperature storage of the infusion solution and the duration of the infusion.
3 Dosage Forms And Strengths ⮝
NEUPOGEN is a clear, colorless, preservative-free solution available as:
Vial:
- Injection: 300 mcg/mL in a single-dose vial
- Injection: 480 mcg/1.6 mL in a single-dose vial
Prefilled Syringe:
- Injection: 300 mcg/0.5 mL in a single-dose prefilled syringe
- Injection: 480 mcg/0.8 mL in a single-dose prefilled syringe
4 Contraindications ⮝
NEUPOGEN is contraindicated in patients with a history of serious allergic reactions to human granulocyte colony-stimulating factors such as filgrastim or pegfilgrastim [see Warnings and Precautions (5.3)].
5 Warnings And Precautions ⮝
5.1 Splenic Rupture
Splenic rupture, including fatal cases, has been reported following the administration of NEUPOGEN. Evaluate patients who report left upper abdominal or shoulder pain for an enlarged spleen or splenic rupture.
5.2 Acute Respiratory Distress Syndrome
Acute respiratory distress syndrome (ARDS) has been reported in patients receiving NEUPOGEN. Evaluate patients who develop fever and lung infiltrates or respiratory distress for ARDS. Discontinue NEUPOGEN in patients with ARDS.
5.3 Serious Allergic Reactions
Serious allergic reactions, including anaphylaxis, have been reported in patients receiving NEUPOGEN. The majority of reported events occurred upon initial exposure. Provide symptomatic treatment for allergic reactions. Allergic reactions, including anaphylaxis, in patients receiving NEUPOGEN can recur within days after the discontinuation of initial anti-allergic treatment. Permanently discontinue NEUPOGEN in patients with serious allergic reactions. NEUPOGEN is contraindicated in patients with a history of serious allergic reactions to human granulocyte colony-stimulating factors such as filgrastim or pegfilgrastim.
5.4 Sickle Cell Disorders
Severe and sometimes fatal sickle cell crises can occur in patients with sickle cell disorders receiving filgrastim products. Discontinue NEUPOGEN if sickle cell crisis occurs.
5.5 Glomerulonephritis
Glomerulonephritis has occurred in patients receiving NEUPOGEN. The diagnoses were based upon azotemia, hematuria (microscopic and macroscopic), proteinuria, and renal biopsy. Generally, events of glomerulonephritis resolved after dose reduction or discontinuation of NEUPOGEN. If glomerulonephritis is suspected, evaluate for cause. If causality is likely, consider dose-reduction or interruption of NEUPOGEN.
5.6 Alveolar Hemorrhage and Hemoptysis
Alveolar hemorrhage manifesting as pulmonary infiltrates and hemoptysis requiring hospitalization have been reported in NEUPOGEN-treated healthy donors undergoing peripheral blood progenitor cell (PBPC) collection mobilization. Hemoptysis resolved with discontinuation of NEUPOGEN. The use of NEUPOGEN for PBPC mobilization in healthy donors is not an approved indication.
5.7 Capillary Leak Syndrome
Capillary leak syndrome (CLS) has been reported after G-CSF administration, including NEUPOGEN, and is characterized by hypotension, hypoalbuminemia, edema and hemoconcentration. Episodes vary in frequency, severity and may be life-threatening if treatment is delayed. Patients who develop symptoms of capillary leak syndrome should be closely monitored and receive standard symptomatic treatment, which may include a need for intensive care.
5.8 Patients with Severe Chronic Neutropenia
Confirm the diagnosis of SCN before initiating NEUPOGEN therapy.
Myelodysplastic syndrome (MDS) and acute myelogenous leukemia (AML) have been reported to occur in the natural history of congenital neutropenia without cytokine therapy. Cytogenetic abnormalities, transformation to MDS, and AML have also been observed in patients treated with NEUPOGEN for SCN. Based on available data including a postmarketing surveillance study, the risk of developing MDS and AML appears to be confined to the subset of patients with congenital neutropenia. Abnormal cytogenetics and MDS have been associated with the eventual development of myeloid leukemia. The effect of NEUPOGEN on the development of abnormal cytogenetics and the effect of continued NEUPOGEN administration in patients with abnormal cytogenetics or MDS are unknown. If a patient with SCN develops abnormal cytogenetics or myelodysplasia the risks and benefits of continuing NEUPOGEN should be carefully considered.
5.9 Thrombocytopenia
Thrombocytopenia has been reported in patients receiving NEUPOGEN. Monitor platelet counts.
5.10 Leukocytosis
Patients with Cancer Receiving Myelosuppressive Chemotherapy
White blood cell counts of 100 000/mm3 or greater were observed in approximately 2% of patients receiving NEUPOGEN at dosages above 5 mcg/kg/day. In patients with cancer receiving NEUPOGEN as an adjunct to myelosuppressive chemotherapy to avoid the potential risks of excessive leukocytosis it is recommended that NEUPOGEN therapy be discontinued if the ANC surpasses 10 000/mm3 after the chemotherapy-induced ANC nadir has occurred. Monitor CBCs at least twice weekly during therapy. Dosages of NEUPOGEN that increase the ANC beyond 10 000/mm3 may not result in any additional clinical benefit. In patients with cancer receiving myelosuppressive chemotherapy discontinuation of NEUPOGEN therapy usually resulted in a 50% decrease in circulating neutrophils within 1 to 2 days with a return to pretreatment levels in 1 to 7 days.
Peripheral Blood Progenitor Cell Collection and Therapy
During the period of administration of NEUPOGEN for PBPC mobilization in patients with cancer, discontinue NEUPOGEN if the leukocyte count rises to > 100,000/mm3.
5.11 Cutaneous Vasculitis
Cutaneous vasculitis has been reported in patients treated with NEUPOGEN. In most cases the severity of cutaneous vasculitis was moderate or severe. Most of the reports involved patients with SCN receiving long-term NEUPOGEN therapy. Hold NEUPOGEN therapy in patients with cutaneous vasculitis. NEUPOGEN may be started at a reduced dose when the symptoms resolve and the ANC has decreased.
5.12 Potential Effect on Malignant Cells
NEUPOGEN is a growth factor that primarily stimulates neutrophils. The granulocyte colony-stimulating factor (G-CSF) receptor through which filgrastim acts has also been found on tumor cell lines. The possibility that filgrastim acts as a growth factor for any tumor type cannot be excluded. The safety of filgrastim in chronic myeloid leukemia (CML) and myelodysplasia has not been established.
When NEUPOGEN is used to mobilize PBPC tumor cells may be released from the marrow and subsequently collected in the leukapheresis product. The effect of reinfusion of tumor cells has not been well studied and the limited data available are inconclusive.
5.13 Simultaneous Use with Chemotherapy and Radiation Therapy Not Recommended
The safety and efficacy of NEUPOGEN given simultaneously with cytotoxic chemotherapy have not been established. Because of the potential sensitivity of rapidly dividing myeloid cells to cytotoxic chemotherapy do not use NEUPOGEN in the period 24 hours before through 24 hours after the administration of cytotoxic chemotherapy [see Dosage and Administration (2.2)].
The safety and efficacy of NEUPOGEN have not been evaluated in patients receiving concurrent radiation therapy. Avoid the simultaneous use of NEUPOGEN with chemotherapy and radiation therapy.
5.14 Nuclear Imaging
Increased hematopoietic activity of the bone marrow in response to growth factor therapy has been associated with transient positive bone-imaging changes. This should be considered when interpreting bone-imaging results.
5.15 Aortitis
Aortitis has been reported in patients receiving NEUPOGEN. It may occur as early as the first week after start of therapy. Manifestations may include generalized signs and symptoms such as fever, abdominal pain, malaise, back pain, and increased inflammatory markers (e.g., c-reactive protein and white blood cell count). Consider aortitis in patients who develop these signs and symptoms without known etiology. Discontinue NEUPOGEN if aortitis is suspected.
6 Adverse Reactions ⮝
The following serious adverse reactions are discussed in greater detail in other sections of the labeling:
- Splenic Rupture [see Warnings and Precautions (5.1)]
- Acute Respiratory Distress Syndrome [see Warnings and Precautions (5.2)]
- Serious Allergic Reactions [see Warnings and Precautions (5.3)]
- Sickle Cell Disorders [see Warnings and Precautions (5.4)]
- Glomerulonephritis [see Warnings and Precautions (5.5)]
- Alveolar Hemorrhage and Hemoptysis [see Warnings and Precautions (5.6)]
- Capillary Leak Syndrome [see Warnings and Precautions (5.7)]
- Thrombocytopenia [see Warnings and Precautions (5.9)]
- Leukocytosis [see Warnings and Precautions (5.10)]
- Cutaneous Vasculitis [see Warnings and Precautions (5.11)]
- Aortitis [see Warnings and Precautions (5.15)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Adverse Reactions in Patients with Cancer Receiving Myelosuppressive Chemotherapy
The following adverse reaction data in Table 2 are from three randomized, placebo-controlled studies in patients with:
- small cell lung cancer receiving standard dose chemotherapy with cyclophosphamide doxorubicin and etoposide (Study 1)
- small cell lung cancer receiving ifosfamide, doxorubicin and etoposide (Study 2), and
- non-Hodgkin s lymphoma (NHL) receiving doxorubicin, cyclophosphamide, vindesine, bleomycin, methylprednisolone, and methotrexate ( ACVBP ) or mitoxantrone, ifosfamide, mitoguazone, teniposide, methotrexate, folinic acid, methylprednisolone, and methotrexate ( VIM3 ) (Study 3).
A total of 451 patients were randomized to receive subcutaneous NEUPOGEN 230 mcg/m2 (Study 1), 240 mcg/m2 (Study 2) or 4 or 5 mcg/kg/day (Study 3) (n = 294) or placebo (n = 157). The patients in these studies were median age 61 (range 29 to 78) years and 64% were male. The ethnicity was 95% Caucasian, 4% African American, and 1% Asian.
Table 2. Adverse Reactions in Patients with Cancer Receiving Myelosuppressive Chemotherapy (With 5% Higher Incidence in NEUPOGEN Compared to Placebo) System Organ Class
Preferred TermNEUPOGEN
(N = 294)
Placebo
(N = 157)
Blood and lymphatic system disorders Thrombocytopenia 38% 29% Gastrointestinal disorders Nausea 43% 32% General disorders and administration site conditions Pyrexia 48% 29% Chest pain 13% 6% Pain 12% 6% Fatigue 20% 10% Musculoskeletal and connective tissue disorders Back pain 15% 8% Arthralgia 9% 2% Bone pain 11% 6% Pain in extremity* 7% 3% Nervous system disorders Dizziness 14% 3% Respiratory, thoracic and mediastinal disorders Cough 14% 8% Dyspnea 13% 8% Skin and subcutaneous tissue disorders Rash 14% 5% Investigations Blood lactate dehydrogenase increased 6% 1% Blood alkaline phosphatase increased 6% 1% * Percent difference (NEUPOGEN Placebo) was 4%.
Adverse events with 5% higher incidence in NEUPOGEN patients compared to placebo and associated with the sequelae of the underlying malignancy or cytotoxic chemotherapy delivered included anemia, constipation, diarrhea, oral pain, vomiting, asthenia, malaise, edema peripheral, hemoglobin decreased, decreased appetite, oropharyngeal pain, and alopecia.
Adverse Reactions in Patients with Acute Myeloid Leukemia
Adverse reaction data below are from a randomized, double-blind, placebo-controlled study in patients with AML (Study 4) who received an induction chemotherapy regimen of intravenous daunorubicin days 1, 2, and 3; cytosine arabinoside days 1 to 7; and etoposide days 1 to 5 and up to 3 additional courses of therapy (induction 2, and consolidation 1, 2) of intravenous daunorubicin, cytosine arabinoside, and etoposide. The safety population included 518 patients randomized to receive either 5 mcg/kg/day NEUPOGEN (n = 257) or placebo (n = 261). The median age was 54 (range 16 to 89) years and 54% were male.
Adverse reactions with 2% higher incidence in NEUPOGEN patients compared to placebo included epistaxis, back pain, pain in extremity, erythema, and rash maculo-papular.
Adverse events with 2% higher incidence in NEUPOGEN patients compared to placebo and associated with the sequelae of the underlying malignancy or cytotoxic chemotherapy included diarrhea, constipation, and transfusion reaction.
Adverse Reactions in Patients with Cancer Undergoing Bone Marrow Transplantation
The following adverse reaction data are from one randomized, no treatment-controlled study in patients with acute lymphoblastic leukemia or lymphoblastic lymphoma receiving high-dose chemotherapy (cyclophosphamide or cytarabine, and melphalan) and total body irradiation (Study 5) and one randomized, no treatment-controlled study in patients with Hodgkin's disease (HD) and NHL undergoing high-dose chemotherapy and autologous bone marrow transplantation (Study 6). Patients receiving autologous bone marrow transplantation only were included in the analysis. A total of 100 patients received either 30 mcg/kg/day as a 4-hour infusion (Study 5) or 10 mcg/kg/day or 30 mcg/kg/day as a 24-hour infusion (Study 6) NEUPOGEN (n = 72), no treatment control or placebo (n = 28). The median age was 30 (range 15 to 57) years, 57% were male.
Adverse reactions with 5% higher incidence in NEUPOGEN patients compared to patients receiving no NEUPOGEN included rash and hypersensitivity.
Adverse reactions in patients receiving intensive chemotherapy followed by autologous BMT with 5% higher incidence in NEUPOGEN patients compared to patients receiving no NEUPOGEN included thrombocytopenia, anemia, hypertension, sepsis, bronchitis, and insomnia.
Adverse Reactions in Patients with Cancer Undergoing Autologous Peripheral Blood Progenitor Cell Collection
The adverse reaction data in Table 3 are from a series of 7 trials in patients with cancer undergoing mobilization of autologous peripheral blood progenitor cells for collection by leukapheresis. Patients (n = 166) in all these trials underwent a similar mobilization/collection regimen: NEUPOGEN was administered for 6 to 8 days in most cases the apheresis procedure occurred on days 5 6, and 7. The dosage of NEUPOGEN ranged between 5 to 30 mcg/kg/day and was administered subcutaneously by injection or continuous infusion. The median age was 39 (range 15 to 67) years, and 48% were male.
Table 3. Adverse Reactions in Patients with Cancer Undergoing Autologous PBPC in the Mobilization Phase ( 5% Incidence in NEUPOGEN Patients) System Organ Class
Preferred TermMobilization Phase
(N = 166)
Musculoskeletal and connective tissue disorders Bone pain 30% General disorders and administration site conditions Pyrexia 16% Investigations Blood alkaline phosphatase increased 11% Nervous system disorders Headache 10% Adverse Reactions in Patients with Severe Chronic Neutropenia
The following adverse reaction data were identified in a randomized, controlled study in patients with SCN receiving NEUPOGEN (Study 7). 123 patients were randomized to a 4-month observation period followed by subcutaneous NEUPOGEN treatment or immediate subcutaneous NEUPOGEN treatment. The median age was 12 years (range 7 months to 76 years) and 46% were male. The dosage of NEUPOGEN was determined by the category of neutropenia. Initial dosage of NEUPOGEN:
- Idiopathic neutropenia: 3.6 mcg/kg/day
- Cyclic neutropenia: 6 mcg/kg/day
- Congenital neutropenia: 6 mcg/kg/day divided 2 times per day
The dosage was increased incrementally to 12 mcg/kg/day divided 2 times per day if there was no response.
Adverse reactions with 5% higher incidence in NEUPOGEN patients compared to patients receiving no NEUPOGEN included arthralgia, bone pain, back pain, muscle spasms, musculoskeletal pain, pain in extremity, splenomegaly, anemia, upper respiratory tract infection, and urinary tract infection (upper respiratory tract infection and urinary tract infection were higher in the NEUPOGEN arm, total infection related events were lower in NEUPOGEN treated patients), epistaxis, chest pain, diarrhea, hypoesthesia, and alopecia.
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay, and the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to filgrastim in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
The incidence of antibody development in patients receiving NEUPOGEN has not been adequately determined. While available data suggest that a small proportion of patients developed binding antibodies to filgrastim, the nature and specificity of these antibodies has not been adequately studied. In clinical studies using NEUPOGEN, the incidence of antibodies binding to filgrastim was 3% (11/333). In these 11 patients, no evidence of a neutralizing response was observed using a cell based bioassay.
Cytopenias resulting from an antibody response to exogenous growth factors have been reported on rare occasions in patients treated with other recombinant growth factors.
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of NEUPOGEN. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- splenic rupture and splenomegaly (enlarged spleen) [see Warnings and Precautions (5.1)]
- acute respiratory distress syndrome [see Warnings and Precautions (5.2)]
- anaphylaxis [see Warnings and Precautions (5.3)]
- sickle cell disorders [see Warnings and Precautions (5.4)]
- glomerulonephritis [see Warnings and Precautions (5.5)]
- alveolar hemorrhage and hemoptysis [see Warnings and Precautions (5.6)]
- capillary leak syndrome [see Warnings and Precautions (5.7)]
- leukocytosis [see Warnings and Precautions (5.10)]
- cutaneous vasculitis [see Warnings and Precautions (5.11)]
- Sweet s syndrome (acute febrile neutrophilic dermatosis)
- decreased bone density and osteoporosis in pediatric patients receiving chronic treatment with NEUPOGEN
- aortitis [see Warnings and Precautions (5.15)]
8 Use In Specific Populations ⮝
8.1 Pregnancy
Risk Summary
Available data from published studies, including several observational studies of pregnancy outcomes in women exposed to filgrastim products and those who were unexposed, have not established an association with NEUPOGEN use during pregnancy and major birth defects, miscarriage, or adverse maternal or fetal outcomes (see Data). Reports in the scientific literature have described transplacental passage of NEUPOGEN in pregnant women when administered 30 hours prior to preterm delivery ( 30 weeks gestation). In animal reproduction studies, effects of filgrastim on prenatal development have been studied in rats and rabbits. No malformations were observed in either species. No maternal or fetal effects were observed in pregnant rats at doses up to 58 times the human doses. Filgrastim has been shown to have adverse effects in pregnant rabbits at doses 2 to 10 times higher than the human doses (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15- 20%, respectively.
Data
Human Data
Several observational studies based on the Severe Chronic Neutropenia International Registry (SCNIR) described pregnancy outcomes in women with severe chronic neutropenia (SCN) who were exposed to filgrastim products during pregnancy and women with SCN who were unexposed. No major differences were seen between treated and untreated women with respect to pregnancy outcome (including miscarriage and preterm labor), newborn complications (including birth weight), and infections. Methodological limitations of these studies include small sample size and lack of generalizability due to the underlying maternal condition.
Animal Data
Effects of filgrastim on prenatal development have been studied in rats and rabbits. No malformations were observed in either species. Filgrastim has been shown to have adverse effects in pregnant rabbits at doses 2 to 10 times higher than the human doses. In pregnant rabbits showing signs of maternal toxicity, reduced embryo-fetal survival (at 20 and 80 mcg/kg/day) and increased abortions (at 80 mcg/kg/day) were observed. In pregnant rats, no maternal or fetal effects were observed at doses up to 575 mcg/kg/day, which is approximately 58 times higher than the human dose of 10 mcg/kg/day.
Offspring of rats administered filgrastim during the peri-natal and lactation periods exhibited a delay in external differentiation and growth retardation ( 20 mcg/kg/day) and slightly reduced survival rate (100 mcg/kg/day).
8.2 Lactation
Risk Summary
There is published literature documenting transfer of filgrastim into human milk. There are a few case reports describing the use of filgrastim in breastfeeding mothers with no adverse effects noted in the infants. There are no data on the effects of filgrastim on milk production. Other filgrastim products are secreted poorly into breast milk, and filgrastim products are not absorbed orally by neonates. The developmental and health benefits of breastfeeding should be considered along with the mother s clinical need for NEUPOGEN and any potential adverse effects on the breastfed child from NEUPOGEN or from the underlying maternal condition.
8.4 Pediatric Use
In patients with cancer receiving myelosuppressive chemotherapy 15 pediatric patients median age 2.6 (range 1.2 to 9.4) years with neuroblastoma were treated with myelosuppressive chemotherapy (cyclophosphamide cisplatin doxorubicin and etoposide) followed by subcutaneous NEUPOGEN at doses of 5, 10, or 15 mcg/kg/day for 10 days (n = 5/dose) (Study 8). The pharmacokinetics of NEUPOGEN in pediatric patients after chemotherapy are similar to those in adults receiving the same weight-normalized doses, suggesting no age-related differences in the pharmacokinetics of NEUPOGEN. In this population NEUPOGEN was well tolerated. There was one report of palpable splenomegaly and one report of hepatosplenomegaly associated with NEUPOGEN therapy; however, the only consistently reported adverse event was musculoskeletal pain which is no different from the experience in the adult population.
The safety and effectiveness of NEUPOGEN have been established in pediatric patients with SCN [see Clinical Studies (14.5)]. In a phase 3 study (Study 7) to assess the safety and efficacy of NEUPOGEN in the treatment of SCN, 123 patients with a median age of 12 years (range 7 months to 76 years) were studied. Of the 123 patients, 12 were infants (7 months to 2 years of age), 49 were children (2 to 12 years of age), and 9 were adolescents (12 to 16 years of age). Additional information is available from a SCN postmarketing surveillance study, which includes long-term follow-up of patients in the clinical studies and information from additional patients who entered directly into the postmarketing surveillance study. Of the 731 patients in the surveillance study, 429 were pediatric patients < 18 years of age (range 0.9 to 17) [see Indications and Usage (1.5), Dosage and Administration (2.6), and Clinical Studies (14.5)].
Long-term follow-up data from the postmarketing surveillance study suggest that height and weight are not adversely affected in patients who received up to 5 years of NEUPOGEN treatment. Limited data from patients who were followed in the phase 3 study for 1.5 years did not suggest alterations in sexual maturation or endocrine function.
Pediatric patients with congenital types of neutropenia (Kostmann s syndrome, congenital agranulocytosis, or Schwachman-Diamond syndrome) have developed cytogenetic abnormalities and have undergone transformation to MDS and AML while receiving chronic NEUPOGEN treatment. The relationship of these events to NEUPOGEN administration is unknown [see Warnings and Precautions (5.8) and Adverse Reactions (6)].
The use of NEUPOGEN to increase survival in pediatric patients acutely exposed to myelosuppressive doses of radiation is based on studies conducted in animals and clinical data supporting the use of NEUPOGEN in other approved indications [see Dosage and Administration (2.1 to 2.4) and Clinical Studies (14.6)].
8.5 Geriatric Use
Among 855 subjects enrolled in 3 randomized, placebo-controlled trials of NEUPOGEN-treated patients receiving myelosuppressive chemotherapy, there were 232 subjects age 65 or older, and 22 subjects age 75 or older. No overall differences in safety or effectiveness were observed between these subjects and younger subjects.
Clinical studies of NEUPOGEN in other approved indications (i.e., BMT recipients, PBPC mobilization, and SCN) did not include sufficient numbers of subjects aged 65 and older to determine whether elderly subjects respond differently from younger subjects.
10 Overdosage ⮝
The maximum tolerated dose of NEUPOGEN has not been determined. In NEUPOGEN clinical trials of patients with cancer receiving myelosuppressive chemotherapy WBC counts > 100 000/mm3 have been reported in less than 5% of patients but were not associated with any reported adverse clinical effects. Patients in the BMT studies received up to 138 mcg/kg/day without toxic effects although there was a flattening of the dose response curve above daily doses of greater than 10 mcg/kg/day.
11 Description ⮝
Filgrastim is a 175 amino acid human granulocyte colony-stimulating factor (G-CSF) manufactured by recombinant DNA technology. Filgrastim is produced by Escherichia coli (E coli) bacteria into which has been inserted the human granulocyte colony-stimulating factor gene. Filgrastim has a molecular weight of 18 800 daltons. The protein has an amino acid sequence that is identical to the natural sequence predicted from human DNA sequence analysis except for the addition of an N-terminal methionine necessary for expression in E coli. Because filgrastim is produced in E coli the product is non-glycosylated and thus differs from G-CSF isolated from a human cell.
NEUPOGEN injection is a sterile clear colorless preservative-free liquid containing filgrastim at a specific activity of 1.0 0.6 x 108 U/mg (as measured by a cell mitogenesis assay). The product is available in single-dose vials and prefilled syringes. The single-dose vials contain either 300 mcg/mL or 480 mcg/1.6 mL of filgrastim. The single-dose prefilled syringes contain either 300 mcg/0.5 mL or 480 mcg/0.8 mL of filgrastim. See table below for product composition of each single-dose vial or prefilled syringe.
300 mcg/mL Vial 480 mcg/1.6 mL Vial 300 mcg/0.5 mL Syringe 480 mcg/0.8 mL Syringe filgrastim 300 mcg 480 mcg 300 mcg 480 mcg acetate 0.59 mg 0.94 mg 0.295 mg 0.472 mg polysorbate 80 0.04 mg 0.064 mg 0.02 mg 0.032 mg sodium 0.035 mg 0.056 mg 0.0175 mg 0.028 mg sorbitol 50 mg 80 mg 25 mg 40 mg water for Injection USP q.s. ad* 1 mL 1.6 mL 0.5 mL 0.8 mL * quantity sufficient to make
12 Clinical Pharmacology ⮝
12.1 Mechanism of Action
Colony-stimulating factors are glycoproteins which act on hematopoietic cells by binding to specific cell surface receptors and stimulating proliferation differentiation commitment and some end-cell functional activation.
Endogenous G-CSF is a lineage-specific colony-stimulating factor that is produced by monocytes fibroblasts, and endothelial cells. G-CSF regulates the production of neutrophils within the bone marrow and affects neutrophil progenitor proliferation differentiation, and selected end-cell functions (including enhanced phagocytic ability priming of the cellular metabolism associated with respiratory burst antibody-dependent killing, and the increased expression of some cell surface antigens). G-CSF is not species-specific and has been shown to have minimal direct in vivo or in vitro effects on the production or activity of hematopoietic cell types other than the neutrophil lineage.
12.2 Pharmacodynamics
In phase 1 studies involving 96 patients with various nonmyeloid malignancies NEUPOGEN administration resulted in a dose-dependent increase in circulating neutrophil counts over the dose range of 1 to 70 mcg/kg/day. This increase in neutrophil counts was observed whether NEUPOGEN was administered intravenous (1 to 70 mcg/kg twice daily) subcutaneous (1 to 3 mcg/kg once daily) or by continuous subcutaneous infusion (3 to 11 mcg/kg/day). With discontinuation of NEUPOGEN therapy neutrophil counts returned to baseline in most cases within 4 days. Isolated neutrophils displayed normal phagocytic (measured by zymosan-stimulated chemoluminescence) and chemotactic (measured by migration under agarose using N-formyl-methionyl-leucyl-phenylalanine [fMLP] as the chemotaxin) activity in vitro.
The absolute monocyte count was reported to increase in a dose-dependent manner in most patients receiving NEUPOGEN; however the percentage of monocytes in the differential count remained within the normal range. Absolute counts of both eosinophils and basophils did not change and were within the normal range following administration of NEUPOGEN. Increases in lymphocyte counts following NEUPOGEN administration have been reported in some normal subjects and patients with cancer.
White blood cell (WBC) differentials obtained during clinical trials have demonstrated a shift towards earlier granulocyte progenitor cells (left shift) including the appearance of promyelocytes and myeloblasts usually during neutrophil recovery following the chemotherapy-induced nadir. In addition Dohle bodies increased granulocyte granulation and hypersegmented neutrophils have been observed. Such changes were transient and were not associated with clinical sequelae, nor were they necessarily associated with infection.
12.3 Pharmacokinetics
Filgrastim exhibits nonlinear pharmacokinetics. Clearance is dependent on filgrastim concentration and neutrophil count: G-CSF receptor-mediated clearance is saturated by high concentration of NEUPOGEN and is diminished by neutropenia. In addition, filgrastim is cleared by the kidney.
Subcutaneous administration of 3.45 mcg/kg and 11.5 mcg/kg of filgrastim resulted in maximum serum concentrations of 4 and 49 ng/mL respectively within 2 to 8 hours. After intravenous administration, the volume of distribution averaged 150 mL/kg and the elimination half-life was approximately 3.5 hours in both normal subjects and cancer subjects. Clearance rates of filgrastim were approximately 0.5 to 0.7 mL/minute/kg. Single parenteral doses or daily intravenous doses over a 14-day period resulted in comparable half-lives. The half-lives were similar for intravenous administration (231 minutes following doses of 34.5 mcg/kg) and for subcutaneous administration (210 minutes following NEUPOGEN dosages of 3.45 mcg/kg). Continuous 24-hour intravenous infusions of 20 mcg/kg over an 11 to 20-day period produced steady-state serum concentrations of filgrastim with no evidence of drug accumulation over the time period investigated. The absolute bioavailability of filgrastim after subcutaneous administration is 60% to 70%.
Specific Populations
Patients Acutely Exposed to Myelosuppressive Doses of Radiation
The pharmacokinetics of filgrastim is not available in patients acutely exposed to myelosuppressive doses of radiation. Based on limited pharmacokinetics data in irradiated non-human primates, the area under the time-concentration curve (AUC), reflecting the exposure to filgrastim in non-human primates at 10 mcg/kg dose of NEUPOGEN, appears to be similar to that in humans at 5 mcg/kg. Simulations conducted using the population pharmacokinetic model indicates that the exposures to filgrastim at a NEUPOGEN dose of 10 mcg/kg in patients acutely exposed to myelosuppressive doses of radiation are expected to exceed the exposures at a dose of 10 mcg/kg in irradiated non-human primates.
Pediatric Patients
The pharmacokinetics of filgrastim in pediatric patients after chemotherapy are similar to those in adult patients receiving the same weight-normalized doses, suggesting no age-related differences in the pharmacokinetics of filgrastim [see Use in Specific Populations (8.4)].
Renal Impairment
In a study with healthy volunteers, subjects with moderate renal impairment, and subjects with end-stage renal disease (n = 4 per group), higher serum concentrations were observed in subjects with end-stage renal disease. However, dose adjustment in patients with renal impairment is not necessary.
Hepatic Impairment
Pharmacokinetics and pharmacodynamics of filgrastim are similar between subjects with hepatic impairment and healthy subjects (n = 12/group). The study included 10 subjects with mild hepatic impairment (Child-Pugh Class A) and 2 subjects with moderate hepatic impairment (Child-Pugh Class B). Therefore, filgrastim dose adjustment for patients with hepatic impairment is not necessary.
13 Nonclinical Toxicology ⮝
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenic potential of filgrastim has not been studied. Filgrastim failed to induce bacterial gene mutations in either the presence or absence of a drug metabolizing enzyme system. Filgrastim had no observed effect on the fertility of male or female rats at doses up to 500 mcg/kg.
13.2 Animal Toxicology and Pharmacology
Filgrastim was administered to monkeys dogs hamsters rats and mice as part of a nonclinical toxicology program, which included studies up to 1-year duration.
In the repeated-dose studies changes observed were attributable to the expected pharmacological actions of filgrastim (i.e. dose-dependent increases in white blood cell counts increased circulating segmented neutrophils and increased myeloid:erythroid ratio in bone marrow). Histopathologic examination of the liver and spleen revealed evidence of ongoing extramedullary granulopoiesis, and dose-related increases in spleen weight were seen in all species. These changes all reversed after discontinuation of treatment.
14 Clinical Studies ⮝
14.1 Patients with Cancer Receiving Myelosuppressive Chemotherapy
The safety and efficacy of NEUPOGEN to decrease the incidence of infection as manifested by febrile neutropenia in patients with nonmyeloid malignancies receiving myelosuppressive anti-cancer drugs were established in a randomized double-blind placebo-controlled trial conducted in patients with small cell lung cancer (Study 1).
In Study 1, patients received up to 6 cycles of intravenous chemotherapy including intravenous cyclophosphamide and doxorubicin on day 1; and etoposide on days 1, 2, and 3 of 21 day cycles. Patients were randomized to receive NEUPOGEN (n = 99) at a dose of 230 mcg/m2 (4 to 8 mcg/kg/day) or placebo (n = 111). Study drug was administered subcutaneously daily beginning on day 4, for a maximum of 14 days. A total of 210 patients were evaluable for efficacy and 207 were evaluable for safety. The demographic and disease characteristics were balanced between arms with a median age of 62 (range 31 to 80) years; 64% males; 89% Caucasian; 72% extensive disease and 28% limited disease.
The main efficacy endpoint was the incidence of febrile neutropenia. Febrile neutropenia was defined as an ANC < 1,000/mm3 and temperature > 38.2 C. Treatment with NEUPOGEN resulted in a clinically and statistically significant reduction in the incidence of infection as manifested by febrile neutropenia, 40% for NEUPOGEN-treated patients and 76% for placebo-treated patients (p < 0.001). There were also statistically significant reductions in the incidence and overall duration of infection manifested by febrile neutropenia; the incidence, severity and duration of severe neutropenia (ANC < 500/mm3); the incidence and overall duration of hospital admissions; and the number of reported days of antibiotic use.
14.2 Patients with Acute Myeloid Leukemia Receiving Induction or Consolidation Chemotherapy
The safety and efficacy of NEUPOGEN to reduce the time to neutrophil recovery and the duration of fever, following induction or consolidation chemotherapy treatment of patients with acute myeloid leukemia (AML) was established in a randomized, double-blind placebo-controlled multi-center trial in patients with newly diagnosed, de novo AML (Study 4).
In Study 4 the initial induction therapy consisted of intravenous daunorubicin days 1, 2, and 3; cytosine arabinoside days 1 to 7; and etoposide days 1 to 5. Patients were randomized to receive subcutaneous NEUPOGEN (n = 259) at a dose of 5 mcg/kg/day or placebo (n = 262) from 24 hours after the last dose of chemotherapy until neutrophil recovery (ANC 1,000/mm3 for 3 consecutive days or 10,000/mm3 for 1 day) or for a maximum of 35 days. The demographic and disease characteristics were balanced between arms with a median age of 54 (range 16 to 89) years; 54% males; initial white blood cell count (65% < 25,000/mm3 and 27% > 100,000/mm3); 29% unfavorable cytogenetics.
The main efficacy endpoint was median duration of severe neutropenia defined as neutrophil count < 500/mm3. Treatment with NEUPOGEN resulted in a clinically and statistically significant reduction in median number of days of severe neutropenia, NEUPOGEN-treated patients 14 days, placebo-treated patients 19 days (p = 0.0001: difference of 5 days (95% CI: -6.0, -4.0)). There was a reduction in the median duration of intravenous antibiotic use, NEUPOGEN-treated patients: 15 days versus placebo-treated patients: 18.5 days; a reduction in the median duration of hospitalization, NEUPOGEN-treated patients: 20 days versus placebo-treated patients: 25 days.
There were no statistically significant differences between the NEUPOGEN and the placebo groups in complete remission rate (69% - NEUPOGEN, 68% - placebo), median time to progression of all randomized patients (165 days - NEUPOGEN, 186 days - placebo), or median overall survival (380 days - NEUPOGEN, 425 days - placebo).
14.3 Patients with Cancer Undergoing Bone Marrow Transplantation
The safety and efficacy of NEUPOGEN to reduce the duration of neutropenia in patients with nonmyeloid malignancies undergoing myeloablative chemotherapy followed by autologous bone marrow transplantation was evaluated in 2 randomized controlled trials of patients with lymphoma (Study 6 and Study 9). The safety and efficacy of NEUPOGEN to reduce the duration of neutropenia in patients undergoing myeloablative chemotherapy followed by allogeneic bone marrow transplantation was evaluated in a randomized placebo-controlled trial (Study 10).
In Study 6, patients with Hodgkin s disease received a preparative regimen of intravenous cyclophosphamide, etoposide, and BCNU ( CVP ), and patients with non-Hodgkin s lymphoma received intravenous BCNU, etoposide, cytosine arabinoside and melphalan ( BEAM ). There were 54 patients randomized 1:1:1 to control, NEUPOGEN 10 mcg/kg/day, and NEUPOGEN 30 mcg/kg/day as a 24-hour continuous infusion starting 24 hours after bone marrow infusion for a maximum of 28 days. The median age was 33 (range 17 to 57) years; 56% males; 69% Hodgkin s disease and 31% non-Hodgkin s lymphoma.
The main efficacy endpoint was duration of severe neutropenia ANC < 500/mm3. A statistically significant reduction in the median number of days of severe neutropenia (ANC < 500/mm3) occurred in the NEUPOGEN-treated groups versus the control group (23 days in the control group 11 days in the 10 mcg/kg/day group, and 14 days in the 30 mcg/kg/day group [11 days in the combined treatment groups p = 0.004]).
In Study 9, patients with Hodgkin s disease and non-Hodgkin s lymphoma received a preparative regimen of intravenous cyclophosphamide, etoposide, and BCNU ( CVP ). There were 43 evaluable patients randomized to continuous subcutaneous infusion NEUPOGEN 10 mcg/kg/day (n = 19), NEUPOGEN 30 mcg/kg/day (n = 10) and no treatment (n = 14) starting the day after marrow infusion for a maximum of 28 days. The median age was 33 (range 17 to 56) years; 67% males; 28% Hodgkin s disease and 72% non-Hodgkin s lymphoma.
The main efficacy endpoint was duration of severe neutropenia. There was statistically significant reduction in the median number of days of severe neutropenia (ANC < 500/mm3) in the NEUPOGEN-treated groups versus the control group (21.5 days in the control group versus 10 days in the NEUPOGEN-treated groups, p < 0.001). The number of days of febrile neutropenia was also reduced significantly in this study (13.5 days in the control group versus 5 days in the NEUPOGEN-treated groups p < 0.0001).
In Study 10, 70 patients scheduled to undergo bone marrow transplantation for multiple underlying conditions using multiple preparative regimens were randomized to receive NEUPOGEN 300 mcg/m2/day (n = 33) or placebo (n = 37) days 5 through 28 after marrow infusion. The median age was 18 (range 1 to 45) years, 56% males. The underlying disease was: 67% hematologic malignancy, 24% aplastic anemia, 9% other. A statistically significant reduction in the median number of days of severe neutropenia occurred in the treated group versus the control group (19 days in the control group and 15 days in the treatment group p < 0.001) and time to recovery of ANC to 500/mm3 (21 days in the control group and 16 days in the treatment group p < 0.001).
14.4 Patients Undergoing Autologous Peripheral Blood Progenitor Cell Collection and Therapy
The safety and efficacy of NEUPOGEN to mobilize autologous peripheral blood progenitor cells for collection by leukapheresis was supported by the experience in uncontrolled trials, and a randomized trial comparing hematopoietic stem cell rescue using NEUPOGEN mobilized autologous peripheral blood progenitor cells to autologous bone marrow (Study 11). Patients in all these trials underwent a similar mobilization/collection regimen: NEUPOGEN was administered for 6 to 7 days in most cases the apheresis procedure occurred on days 5 6, and 7. The dose of NEUPOGEN ranged between 10 to 24 mcg/kg/day and was administered subcutaneously by injection or continuous intravenous infusion.
Engraftment was evaluated in 64 patients who underwent transplantation using NEUPOGEN mobilized autologous hematopoietic progenitor cells in uncontrolled trials. Two of the 64 patients (3%) did not achieve the criteria for engraftment as defined by a platelet count 20 000/mm3 by day 28. In clinical trials of NEUPOGEN for the mobilization of hematopoietic progenitor cells NEUPOGEN was administered to patients at doses between 5 to 24 mcg/kg/day after reinfusion of the collected cells until a sustainable ANC ( 500/mm3) was reached. The rate of engraftment of these cells in the absence of NEUPOGEN post transplantation has not been studied.
Study 11 was a randomized, unblinded study of patients with Hodgkin s disease or non-Hodgkin s lymphoma undergoing myeloablative chemotherapy 27 patients received NEUPOGEN-mobilized autologous hematopoietic progenitor cells and 31 patients received autologous bone marrow. The preparative regimen was intravenous BCNU, etoposide, cytosine arabinoside and melphalan ( BEAM ). Patients received daily NEUPOGEN 24 hours after stem cell infusion at a dose of 5 mcg/kg/day. The median age was 33 (range 1 to 59) years; 64% males; 57% Hodgkin s disease and 43% non-Hodgkin s lymphoma. The main efficacy endpoint was number of days of platelet transfusions. Patients randomized to NEUPOGEN-mobilized autologous peripheral blood progenitor cells compared to autologous bone marrow had significantly fewer days of platelet transfusions (median 6 vs 10 days).
14.5 Patients with Severe Chronic Neutropenia
The safety and efficacy of NEUPOGEN to reduce the incidence and duration of sequelae of neutropenia (that is fever infections, oropharyngeal ulcers) in symptomatic adult and pediatric patients with congenital neutropenia cyclic neutropenia or idiopathic neutropenia was established in a randomized controlled trial conducted in patients with severe neutropenia (Study 7).
Patients eligible for Study 7 had a history of severe chronic neutropenia documented with an ANC < 500/mm3 on three occasions during a 6-month period, or in patients with cyclic neutropenia 5 consecutive days of ANC < 500/mm3 per cycle. In addition, patients must have experienced a clinically significant infection during the previous 12 months. Patients were randomized to a 4-month observation period followed by NEUPOGEN treatment or immediate NEUPOGEN treatment. The median age was 12 years (range 7 months to 76 years); 46% males; 34% idiopathic, 17% cyclic and 49% congenital neutropenia.
NEUPOGEN was administered subcutaneously. The dose of NEUPOGEN was determined by the category of neutropenia. Initial dose of NEUPOGEN:
- Idiopathic neutropenia: 3.6 mcg/kg/day
- Cyclic neutropenia: 6 mcg/kg/day
- Congenital neutropenia: 6 mcg/kg/day divided 2 times per day
The dose was increased incrementally to 12 mcg/kg/day divided 2 times per day if there was no response.
The main efficacy endpoint was response to NEUPOGEN treatment. ANC response from baseline (< 500/mm3) was defined as follows:
- Complete response: median ANC > 1,500/mm3
- Partial response: median ANC 500/mm3 and 1,500/mm3 with a minimum increase of 100%
- No response: median ANC < 500/mm3
There were 112 of 123 patients who demonstrated a complete or partial response to NEUPOGEN treatment.
Additional efficacy endpoints included a comparison between patients randomized to 4 months of observation and patients receiving NEUPOGEN of the following parameters:
- incidence of infection
- incidence of fever
- duration of fever
- incidence, duration, and severity of oropharyngeal ulcers
- number of days of antibiotic use
The incidence for each of these 5 clinical parameters was lower in the NEUPOGEN arm compared to the control arm for cohorts in each of the 3 major diagnostic categories. An analysis of variance showed no significant interaction between treatment and diagnosis suggesting that efficacy did not differ substantially in the different diseases. Although NEUPOGEN substantially reduced neutropenia in all patient groups in patients with cyclic neutropenia cycling persisted but the period of neutropenia was shortened to 1 day.
14.6 Patients Acutely Exposed to Myelosuppressive Doses of Radiation (Hematopoietic Syndrome of Acute Radiation Syndrome)
Efficacy studies of NEUPOGEN could not be conducted in humans with acute radiation syndrome for ethical and feasibility reasons. Approval of this indication was based on efficacy studies conducted in animals and data supporting the use of NEUPOGEN for other approved indications [see Dosage and Administration (2.1 to 2.4)].
Because of the uncertainty associated with extrapolating animal efficacy data to humans, the selection of human dose for NEUPOGEN is aimed at providing exposures to filgrastim that exceed those observed in animal efficacy studies. The 10 mcg/kg daily dose is selected for humans exposed to myelosuppressive doses of radiation because the exposure associated with such a dose is expected to exceed the exposure associated with a 10 mcg/kg dose in non-human primates [see Pharmacokinetics (12.3)]. The safety of NEUPOGEN at a daily dose of 10 mcg/kg has been assessed on the basis of clinical experience in approved indications.
The efficacy of NEUPOGEN was studied in a randomized, blinded, placebo-controlled study in a non-human primate model of radiation injury. The planned sample size was 62 animals, but the study was stopped at the interim analysis with 46 animals because efficacy was established. Rhesus macaques were randomized to a control (n = 22) or treated (n = 24) group. Animals were exposed to total body irradiation of 7.4 0.15 Gy delivered at 0.8 0.03 Gy/min, representing a dose that would be lethal in 50% of animals by 60 days of follow-up (LD50/60). Starting on day 1 after irradiation, animals received daily subcutaneous injections of placebo (5% dextrose in water) or filgrastim (10 mcg/kg/day). Blinded treatment was stopped when one of the following criteria was met: ANC 1,000/mm3 for 3 consecutive days, or ANC 10,000/mm3 for more than 2 consecutive days within study day 1 to 5, or ANC 10,000/mm3 any time after study day 5. Animals received medical management consisting of intravenous fluids, antibiotics, blood transfusions, and other support as required.
Filgrastim significantly (at 0.023 level of significance) reduced 60-day mortality in the irradiated non-human primates: 21% mortality (5/24) in the filgrastim group compared to 59% mortality (13/22) in the control group.
16 How Supplied/storage And Handling ⮝
NEUPOGEN injection is a clear, colorless, preservative-free solution supplied as:
Vials
Single-dose vials containing 300 mcg/mL of filgrastim. Dispensing packs of 10 vials (NDC 55513-530-10).
Single-dose vials containing 480 mcg/1.6 mL (300 mcg/mL) of filgrastim. Dispensing packs of 10 vials (NDC 55513-546-10).
Prefilled Syringes (SingleJect )
Single-dose, prefilled syringe with 27 gauge, inch needle with an UltraSafe Needle Guard, containing 300 mcg/0.5 mL of filgrastim.
- Pack of 1 prefilled syringe (NDC 55513-924-91).
- Pack of 10 prefilled syringes (NDC 55513-924-10).
Single-dose, prefilled syringe with 27 gauge, inch needle with an UltraSafe Needle Guard, containing 480 mcg/0.8 mL of filgrastim.
- Pack of 1 prefilled syringe (NDC 55513-209-91).
- Pack of 10 prefilled syringes (NDC 55513-209-10).
The needle cap of the prefilled syringe contains dry natural rubber (a derivative of latex) [see Dosage and Administration (2.6)].
Store NEUPOGEN at 2 to 8 C (36 to 46 F) in the carton to protect from light. Do not leave NEUPOGEN in direct sunlight. Avoid freezing; if frozen, thaw in the refrigerator before administration. Discard NEUPOGEN if frozen more than once. Avoid shaking. Transport via a pneumatic tube has not been studied.
No Title 1572449134 ⮝
Patient Information
NEUPOGEN (nu-po-jen)
(filgrastim)
injectionWhat is NEUPOGEN?
NEUPOGEN is a man-made form of granulocyte colony-stimulating factor (G-CSF). G-CSF is a substance produced by the body. It stimulates the growth of neutrophils, a type of white blood cell important in the body s fight against infection.
Acute Radiation Syndrome: The effectiveness of NEUPOGEN for this use was only studied in animals, because it could not be studied in people.Do not take NEUPOGEN if you have had a serious allergic reaction to human G-CSFs such as filgrastim or pegfilgrastim products. Before you take NEUPOGEN, tell your healthcare provider about all of your medical conditions, including if you: Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
- have a sickle cell disorder.
- have kidney problems.
- are receiving radiation therapy.
- are allergic to latex. The needle cap on the prefilled syringe contains dry natural rubber (derived from latex). You should not give NEUPOGEN using the prefilled syringe if you have latex allergies. Ask your healthcare provider about using the vial if you have latex allergies.
- are pregnant or plan to become pregnant. It is not known if NEUPOGEN will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if NEUPOGEN passes into your breast milk.
How will I receive NEUPOGEN?
- NEUPOGEN injections can be given by a healthcare provider by intravenous (IV) infusion or under your skin (subcutaneous injection). Your healthcare provider may decide subcutaneous injections can be given at home by you or your caregiver. If NEUPOGEN is given at home, see the detailed Instructions for Use that comes with your NEUPOGEN for information on how to prepare and inject a dose of NEUPOGEN.
- You and your caregiver should be shown how to prepare and inject NEUPOGEN before you use it, by your healthcare provider.
- Your healthcare provider will tell you how much NEUPOGEN to inject and when to inject it. Do not change your dose or stop NEUPOGEN unless your healthcare provider tells you to.
- If you are receiving NEUPOGEN because you are also receiving chemotherapy, your dose of NEUPOGEN should be injected at least 24 hours before or 24 hours after your dose of chemotherapy. Your healthcare provider will do blood tests to monitor your white blood cell count, and if necessary, adjust your NEUPOGEN dose.
- If you are receiving NEUPOGEN because you have been suddenly (acutely) exposed to an amount of radiation that can affect your bone marrow (Acute Radiation Syndrome), you will need to have blood tests about every 3 days during treatment with NEUPOGEN to check your white blood cell count.
- If you miss a dose of NEUPOGEN, talk to your healthcare provider about when you should give your next dose.
What are the possible side effects of NEUPOGEN?
NEUPOGEN may cause serious side effects, including:The most common side effects experienced in patients receiving NEUPOGEN include:
- Spleen rupture. Your spleen may become enlarged and can rupture. A ruptured spleen can cause death. Call your healthcare provider right away if you have pain in the left upper stomach (abdomen) area or your left shoulder.
- A serious lung problem called acute respiratory distress syndrome (ARDS). Call your healthcare provider or get emergency medical help right away if you have shortness of breath with or without a fever, trouble breathing, or a fast rate of breathing.
- Serious allergic reactions. NEUPOGEN can cause serious allergic reactions. These reactions can cause a rash over your whole body, shortness of breath, wheezing, dizziness, swelling around your mouth or eyes, fast heart rate, and sweating. If you have any of these symptoms, stop using NEUPOGEN and call your healthcare provider or get emergency medical help right away.
- Sickle cell crises. You may have a serious sickle cell crisis, which could lead to death, if you have a sickle cell disorder and receive NEUPOGEN. Call your healthcare provider right away if you have symptoms of sickle cell crisis such as pain or difficulty breathing.
- Kidney injury (glomerulonephritis). NEUPOGEN can cause kidney injury. Call your healthcare provider right away if you develop any of the following symptoms:
swelling of your face or ankles
blood in your urine or dark colored urine
you urinate less than usual
- Capillary leak syndrome. NEUPOGEN can cause fluid to leak from blood vessels into your body s tissues. This condition is called Capillary Leak Syndrome (CLS). CLS can quickly cause you to have symptoms that may become life-threatening. Get emergency medical help right away if you develop any of the following symptoms:
swelling or puffiness and are urinating less than usual
trouble breathing
swelling of your stomach area (abdomen) and feeling of fullness
dizziness or feeling faint
a general feeling of tiredness
- Decreased platelet count (thrombocytopenia). Your healthcare provider will check your blood during treatment with NEUPOGEN. Tell your healthcare provider if you have unusual bleeding or bruising during treatment with NEUPOGEN. This could be a sign of decreased platelet counts, which may reduce the ability of your blood to clot.
- Increased white blood cell count (leukocytosis). Your healthcare provider will check your blood during treatment with NEUPOGEN.
- Inflammation of your blood vessels (cutaneous vasculitis). Tell your healthcare provider right away if you develop purple spots or redness of your skin.
- Inflammation of the aorta (aortitis). Inflammation of the aorta (the large blood vessel which transports blood from the heart to the body) has been reported in patients who received NEUPOGEN. Symptoms may include fever, abdominal pain, feeling tired, and back pain. Call your healthcare provider if you experience these symptoms.
These are not all the possible side effects of NEUPOGEN. Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
- Patients with cancer receiving chemotherapy: fever, pain, rash, cough, and shortness of breath
- Patients with acute myeloid leukemia receiving chemotherapy: pain, nose bleed, and rash
- Patients with cancer receiving chemotherapy followed by bone marrow transplant: rash
- Patients who are having their own blood cells collected: bone pain, fever, and headache
- Patients with severe chronic neutropenia: pain, decreased red blood cells, nose bleed, diarrhea, reduced sensation, and hair loss
How should I store NEUPOGEN? Keep NEUPOGEN out of the reach of children.
- Store NEUPOGEN in the refrigerator between 36 F to 46 F (2 C to 8 C).
- Do not freeze.
- Keep NEUPOGEN in the original carton to protect from light or physical damage. Do not leave NEUPOGEN in direct sunlight.
- Do not shake NEUPOGEN.
- Take NEUPOGEN out of the refrigerator 30 minutes before use and allow it to reach room temperature before preparing an injection.
- Throw away (dispose of) any NEUPOGEN that has been left at room temperature for longer than 24 hours.
- After you inject your dose, throw away (dispose of) any unused NEUPOGEN left in the vials or prefilled syringes. Do not save unused NEUPOGEN in the vials or prefilled syringes for later use.
General information about the safe and effective use of NEUPOGEN.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use NEUPOGEN for a condition for which it was not prescribed. Do not give NEUPOGEN to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about NEUPOGEN that is written for healthcare professionals.What are the ingredients in NEUPOGEN?
Active ingredient: filgrastim
Inactive ingredients: acetate, polysorbate 80, sodium, sorbitol, and water for Injection
Amgen Inc., One Amgen Center Drive, Thousand Oaks, California 91320-1799 U.S.A.
US License No. 1080
Patent: http://pat.amgen.com/neupogen/
1991-2018 Amgen Inc. All rights reserved.
1xxxxxx v16This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 06/2018
Principal Display Panel ⮝
NDC 55513-209-10
10 x 480 mcg/0.8 mL Single Dose Prefilled Syringes with 27 Gauge Needles
AMGEN
Neupogen SingleJect
(filgrastim) injection
A Recombinant Granulocyte Colony Stimulating Factor (rG-CSF) derived from E Coli
480 mcg
Single Dose Prefilled Syringes with 27 Gauge Needles
480 mcg/0.8 mL
For Subcutaneous or Intravenous Use Only
This Product Contains Dry Natural Rubber
Sterile Solution No Preservative
Refrigerate at 2 to 8 C (36 to 46 F). Avoid Shaking.
Rx Only
Manufactured by Amgen Inc.
Thousand Oaks, CA 91320 U.S.A.
U.S. License No. 1080
NEUPOGEN
filgrastim injection, solution
Product Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55513-530 Route of Administration INTRAVENOUS, SUBCUTANEOUS
Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FILGRASTIM (UNII: PVI5M0M1GW) (FILGRASTIM - UNII:PVI5M0M1GW) FILGRASTIM 300 ug in 1 mL
Inactive Ingredients Ingredient Name Strength ACETIC ACID (UNII: Q40Q9N063P) 0.59 mg in 1 mL SORBITOL (UNII: 506T60A25R) 50 mg in 1 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.04 mg in 1 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R)
Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55513-530-10 10 in 1 PACKAGE 05/19/1997 1 NDC:55513-530-01 1 mL in 1 VIAL; Type 0: Not a Combination Product
Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103353 05/19/1997
NEUPOGEN
filgrastim injection, solution
Product Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55513-546 Route of Administration INTRAVENOUS, SUBCUTANEOUS
Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FILGRASTIM (UNII: PVI5M0M1GW) (FILGRASTIM - UNII:PVI5M0M1GW) FILGRASTIM 480 ug in 1.6 mL
Inactive Ingredients Ingredient Name Strength ACETIC ACID (UNII: Q40Q9N063P) 0.94 mg in 1.6 mL SORBITOL (UNII: 506T60A25R) 80 mg in 1.6 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.06 mg in 1.6 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R)
Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55513-546-10 10 in 1 BOX 04/07/1997 1 NDC:55513-546-01 1.6 mL in 1 VIAL; Type 0: Not a Combination Product
Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103353 04/07/1997
NEUPOGEN
filgrastim injection, solution
Product Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55513-924 Route of Administration INTRAVENOUS, SUBCUTANEOUS
Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FILGRASTIM (UNII: PVI5M0M1GW) (FILGRASTIM - UNII:PVI5M0M1GW) FILGRASTIM 300 ug in 0.5 mL
Inactive Ingredients Ingredient Name Strength ACETIC ACID (UNII: Q40Q9N063P) 0.29 mg in 0.5 mL SORBITOL (UNII: 506T60A25R) 25 mg in 0.5 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.02 mg in 0.5 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R)
Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55513-924-10 10 in 1 BOX 10/02/2000 1 NDC:55513-924-01 0.5 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 2 NDC:55513-924-91 0.5 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 10/02/2000
Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103353 10/02/2000
NEUPOGEN
filgrastim injection, solution
Product Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55513-209 Route of Administration INTRAVENOUS, SUBCUTANEOUS
Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FILGRASTIM (UNII: PVI5M0M1GW) (FILGRASTIM - UNII:PVI5M0M1GW) FILGRASTIM 480 ug in 0.8 mL
Inactive Ingredients Ingredient Name Strength ACETIC ACID (UNII: Q40Q9N063P) 0.47 mg in 0.8 mL SORBITOL (UNII: 506T60A25R) 40 mg in 0.8 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.03 mg in 0.8 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R)
Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55513-209-10 10 in 1 BOX 10/02/2000 1 NDC:55513-209-01 0.8 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 2 NDC:55513-209-91 0.8 mL in 1 SYRINGE; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 10/02/2000
Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103353 10/02/2000
Labeler - Amgen Inc (039976196)
Establishment Name Address ID/FEI Business Operations Amgen Manufacturing Ltd 785800020 ANALYSIS(55513-530, 55513-546, 55513-924, 55513-209) , LABEL(55513-530, 55513-546, 55513-924, 55513-209) , MANUFACTURE(55513-530, 55513-546, 55513-924, 55513-209) , PACK(55513-530, 55513-546, 55513-924, 55513-209) Revised: 6/2018 Document Id: 6c14d827-1d18-4657-80ef-3da115da7e2d 34391-3 Set id: 97cc73cc-b5b7-458a-a933-77b00523e193 Version: 150 Effective Time: 20180618 Amgen Inc
Description ⮝
Filgrastim is a human granulocyte colony-stimulating factor (G-CSF) produced by recombinant DNA technology. NEUPOGEN is the Amgen Inc. trademark for Filgrastim which has been selected as the name for recombinant methionyl human granulocyte colony-stimulating factor (r-metHuG-CSF).
NEUPOGEN is a 175 amino acid protein manufactured by recombinant DNA technology.1 NEUPOGEN is produced by Escherichia coli (E coli) bacteria into which has been inserted the human granulocyte colony-stimulating factor gene. NEUPOGEN has a molecular weight of 18 800 daltons. The protein has an amino acid sequence that is identical to the natural sequence predicted from human DNA sequence analysis except for the addition of an N-terminal methionine necessary for expression in E coli. Because NEUPOGEN is produced in E coli the product is nonglycosylated and thus differs from G-CSF isolated from a human cell.
NEUPOGEN is a sterile clear colorless preservative-free liquid for parenteral administration containing Filgrastim at a specific activity of 1.0 0.6 x 108 U/mg (as measured by a cell mitogenesis assay). The product is available in single use vials and prefilled syringes. The single use vials contain either 300 mcg or 480 mcg Filgrastim at a fill volume of 1.0 mL or 1.6 mL, respectively. The single use prefilled syringes contain either 300 mcg or 480 mcg Filgrastim at a fill volume of 0.5 mL or 0.8 mL, respectively. See table below for product composition of each single use vial or prefilled syringe.
300 mcg/
1.0 mL Vial480 mcg/
1.6 mL Vial300 mcg/
0.5 mL Syringe480 mcg/
0.8 mL SyringeFilgrastim 300 mcg 480 mcg 300 mcg 480 mcg Acetate 0.59 mg 0.94 mg 0.295 mg 0.472 mg Sorbitol 50.0 mg 80.0 mg 25.0 mg 40.0 mg Polysorbate 80 0.04 mg 0.064 mg 0.02 mg 0.032 mg Sodium 0.035 mg 0.056 mg 0.0175 mg 0.028 mg Water for Injection USP q.s. ad 1.0 mL 1.6 mL 0.5 mL 0.8 mL
Clinical Pharmacology ⮝
Colony-stimulating FactorsColony-stimulating factors are glycoproteins which act on hematopoietic cells by binding to specific cell surface receptors and stimulating proliferation differentiation commitment and some end-cell functional activation.
Endogenous G-CSF is a lineage specific colony-stimulating factor which is produced by monocytes fibroblasts, and endothelial cells. G-CSF regulates the production of neutrophils within the bone marrow and affects neutrophil progenitor proliferation 2 3 differentiation,2 4 and selected end-cell functional activation (including enhanced phagocytic ability 5 priming of the cellular metabolism associated with respiratory burst 6 antibody dependent killing,7 and the increased expression of some functions associated with cell surface antigens8). G-CSF is not species specific and has been shown to have minimal direct in vivo or in vitro effects on the production of hematopoietic cell types other than the neutrophil lineage.
Preclinical ExperienceFilgrastim was administered to monkeys dogs hamsters rats and mice as part of a preclinical toxicology program which included single-dose acute repeated-dose subacute subchronic and chronic studies. Single-dose administration of Filgrastim by the oral intravenous (IV) subcutaneous (SC) or intraperitoneal (IP) routes resulted in no significant toxicity in mice rats hamsters or monkeys. Although no deaths were observed in mice rats or monkeys at dose levels up to 3450 mcg/kg or in hamsters using single doses up to approximately 860 mcg/kg deaths were observed in a subchronic (13-week) study in monkeys. In this study evidence of neurological symptoms was seen in monkeys treated with doses of Filgrastim greater than 1150 mcg/kg/day for up to 18 days. Deaths were seen in 5 of the 8 treated animals and were associated with 15- to 28-fold increases in peripheral leukocyte counts and neutrophil-infiltrated hemorrhagic foci were seen in both the cerebrum and cerebellum. In contrast no monkeys died following 13 weeks of daily IV administration of Filgrastim at a dose level of 115 mcg/kg. In an ensuing 52-week study one 115 mcg/kg dosed female monkey died after 18 weeks of daily IV administration of Filgrastim. Death was attributed to cardiopulmonary insufficiency.
In subacute repeated-dose studies changes observed were attributable to the expected pharmacological actions of Filgrastim (ie dose-dependent increases in white cell counts increased circulating segmented neutrophils and increased myeloid:erythroid ratio in bone marrow). In all species histopathologic examination of the liver and spleen revealed evidence of ongoing extramedullary granulopoiesis; increased spleen weights were seen in all species and appeared to be dose-related. A dose-dependent increase in serum alkaline phosphatase was observed in rats and may reflect increased activity of osteoblasts and osteoclasts. Changes in serum chemistry values were reversible following discontinuation of treatment.
In rats treated at doses of 1150 mcg/kg/day for 4 weeks (5 of 32 animals) and for 13 weeks at doses of 100 mcg/kg/day (4 of 32 animals) and 500 mcg/kg/day (6 of 32 animals) articular swelling of the hind legs was observed. Some degree of hind leg dysfunction was also observed; however symptoms reversed following cessation of dosing. In rats osteoclasis and osteoanagenesis were found in the femur humerus coccyx and hind legs (where they were accompanied by synovitis) after IV treatment for 4 weeks (115 to 1150 mcg/kg/day) and in the sternum after IV treatment for 13 weeks (115 to 575 mcg/kg/day). These effects reversed to normal within 4 to 5 weeks following cessation of treatment.
In the 52-week chronic repeated-dose studies performed in rats (IP injection up to 57.5 mcg/kg/day) and cynomolgus monkeys (IV injection of up to 115 mcg/kg/day) changes observed were similar to those noted in the subacute studies. Expected pharmacological actions of Filgrastim included dose-dependent increases in white cell counts increased circulating segmented neutrophils and alkaline phosphatase levels and increased myeloid:erythroid ratios in the bone marrow. Decreases in platelet counts were also noted in primates. In no animals tested were hemorrhagic complications observed. Rats displayed dose-related swelling of the hind limb accompanied by some degree of hind limb dysfunction; osteopathy was noted microscopically. Enlarged spleens (both species) and livers (monkeys) reflective of ongoing extramedullary granulopoiesis as well as myeloid hyperplasia of the bone marrow were observed in a dose-dependent manner.
Pharmacologic Effects of NEUPOGENIn phase 1 studies involving 96 patients with various nonmyeloid malignancies NEUPOGEN administration resulted in a dose-dependent increase in circulating neutrophil counts over the dose range of 1 to 70 mcg/kg/day.9-11 This increase in neutrophil counts was observed whether NEUPOGEN was administered IV (1 to 70 mcg/kg twice daily) 9 SC (1 to 3 mcg/kg once daily) 11 or by continuous SC infusion (3 to 11 mcg/kg/day).10 With discontinuation of NEUPOGEN therapy neutrophil counts returned to baseline in most cases within 4 days. Isolated neutrophils displayed normal phagocytic (measured by zymosan-stimulated chemoluminescence) and chemotactic (measured by migration under agarose using N-formyl-methionyl-leucyl-phenylalanine [fMLP] as the chemotaxin) activity in vitro.
The absolute monocyte count was reported to increase in a dose-dependent manner in most patients receiving NEUPOGEN ; however the percentage of monocytes in the differential count remained within the normal range. In all studies to date absolute counts of both eosinophils and basophils did not change and were within the normal range following administration of NEUPOGEN . Increases in lymphocyte counts following NEUPOGEN administration have been reported in some normal subjects and cancer patients.
White blood cell (WBC) differentials obtained during clinical trials have demonstrated a shift towards earlier granulocyte progenitor cells (left shift) including the appearance of promyelocytes and myeloblasts usually during neutrophil recovery following the chemotherapy-induced nadir. In addition Dohle bodies increased granulocyte granulation and hypersegmented neutrophils have been observed. Such changes were transient and were not associated with clinical sequelae, nor were they necessarily associated with infection.
PharmacokineticsAbsorption and clearance of NEUPOGEN follows first-order pharmacokinetic modeling without apparent concentration dependence. A positive linear correlation occurred between the parenteral dose and both the serum concentration and area under the concentration-time curves. Continuous IV infusion of 20 mcg/kg of NEUPOGEN over 24 hours resulted in mean and median serum concentrations of approximately 48 and 56 ng/mL respectively. Subcutaneous administration of 3.45 mcg/kg and 11.5 mcg/kg resulted in maximum serum concentrations of 4 and 49 ng/mL respectively within 2 to 8 hours. The volume of distribution averaged 150 mL/kg in both normal subjects and cancer patients. The elimination half-life in both normal subjects and cancer patients was approximately 3.5 hours. Clearance rates of NEUPOGEN were approximately 0.5 to 0.7 mL/minute/kg. Single parenteral doses or daily IV doses over a 14-day period resulted in comparable half-lives. The half-lives were similar for IV administration (231 minutes following doses of 34.5 mcg/kg) and for SC administration (210 minutes following NEUPOGEN doses of 3.45 mcg/kg). Continuous 24-hour IV infusions of 20 mcg/kg over an 11- to 20-day period produced steady-state serum concentrations of NEUPOGEN with no evidence of drug accumulation over the time period investigated.
Pharmacokinetic data in geriatric patients ( 65 years) are not available.
Clinical Experience ⮝
Cancer Patients Receiving Myelosuppressive ChemotherapyNEUPOGEN has been shown to be safe and effective in accelerating the recovery of neutrophil counts following a variety of chemotherapy regimens. In a phase 3 clinical trial in small cell lung cancer patients received SC administration of NEUPOGEN (4 to 8 mcg/kg/day days 4 to 17) or placebo. In this study the benefits of NEUPOGEN therapy were shown to be prevention of infection as manifested by febrile neutropenia decreased hospitalization and decreased IV antibiotic usage. No difference in survival or disease progression was demonstrated.
In the phase 3 randomized double-blind placebo-controlled trial conducted in patients with small cell lung cancer patients were randomized to receive NEUPOGEN (n = 99) or placebo (n = 111) starting on day 4 after receiving standard dose chemotherapy with cyclophosphamide doxorubicin and etoposide. A total of 210 patients were evaluated for efficacy and 207 evaluated for safety. Treatment with NEUPOGEN resulted in a clinically and statistically significant reduction in the incidence of infection as manifested by febrile neutropenia; the incidence of at least one infection over all cycles of chemotherapy was 76% (84/111) for placebo-treated patients versus 40% (40/99) for NEUPOGEN -treated patients (p less than 0.001). The following secondary analyses were also performed. The requirements for in-patient hospitalization and antibiotic use were also significantly decreased during the first cycle of chemotherapy; incidence of hospitalization was 69% (77/111) for placebo-treated patients in cycle 1 versus 52% (51/99) for NEUPOGEN -treated patients (p = 0.032). The incidence of IV antibiotic usage was 60% (67/111) for placebo-treated patients in cycle 1 versus 38% (38/99) for NEUPOGEN -treated patients (p = 0.003). The incidence severity and duration of severe neutropenia (absolute neutrophil count [ANC] less than 500/mm3) following chemotherapy were all significantly reduced. The incidence of severe neutropenia in cycle 1 was 84% (83/99) for patients receiving NEUPOGEN versus 96% (106/110) for patients receiving placebo (p = 0.004). Over all cycles patients randomized to NEUPOGEN had a 57% (286/500 cycles) rate of severe neutropenia versus 77% (416/543 cycles) for patients randomized to placebo. The median duration of severe neutropenia in cycle 1 was reduced from 6 days (range 0 to 10 days) for patients receiving placebo to 2 days (range 0 to 9 days) for patients receiving NEUPOGEN (p less than 0.001). The mean duration of neutropenia in cycle 1 was 5.64 2.27 days for patients receiving placebo versus 2.44 1.90 days for patients receiving NEUPOGEN . Over all cycles the median duration of neutropenia was 3 days for patients randomized to placebo versus 1 day for patients randomized to NEUPOGEN . The median severity of neutropenia (as measured by ANC nadir) was 72/mm3 (range 0/mm3 to 7912/mm3) in cycle 1 for patients receiving NEUPOGEN versus 38/mm3 (range 0/mm3 to 9520/mm3) for patients receiving placebo (p = 0.012). The mean severity of neutropenia in cycle 1 was 496/mm3 1382/mm3 for patients receiving NEUPOGEN versus 204/mm3 953/mm3 for patients receiving placebo. Over all cycles the ANC nadir for patients randomized to NEUPOGEN was 403/mm3 versus 161/mm3 for patients randomized to placebo. Administration of NEUPOGEN resulted in an earlier ANC nadir following chemotherapy than was experienced by patients receiving placebo (day 10 vs day 12). NEUPOGEN was well tolerated when given SC daily at doses of 4 to 8 mcg/kg for up to 14 consecutive days following each cycle of chemotherapy (see ADVERSE REACTIONS).
Several other phase 1/2 studies which did not directly measure the incidence of infection but which did measure increases in neutrophils support the efficacy of NEUPOGEN . The regimens are presented to provide some background on the clinical experience with NEUPOGEN . No claim regarding the safety or efficacy of the chemotherapy regimens is made. The effects of NEUPOGEN on tumor growth or on the anti-tumor activity of the chemotherapy were not assessed. The doses of NEUPOGEN used in these studies are considerably greater than those found to be effective in the phase 3 study described above. Such phase 1/2 studies are summarized in the following table.
Type of
MalignacyRegimen Chemotherapy Dose No. Pts. Trial
PhaseNEUPOGEN Daily Dosage* Small Cell
Lung Cancer
Cyclophosphamide
1 g/m2/day 210
3
4 - 8 mcg/kg SC
Doxorubicin
50 mg/m2/day days 4 - 17
Etoposide
120 mg/m2/day x 3
q 21 days
Small Cell
Lung Cancer11 Ifosfamide
5 g/m2/day 12
1/2
5.75 - 46 mcg/kg IV
Doxorubicin
50 mg/m2/day days 4 - 17
Etoposide
120 mg/m2/day x 3
Mesna
8 g/m2/day q 21 days
Urothelial
|
Cancer12 Methotrexate
30 mg/m2/day x 2
40
1/2
3.45 - 69 mcg/kg IV
Vinblastine
3 mg/m2/day x 2
days 4 - 11
Doxorubicin
30 mg/m2/day Cisplatin
70 mg/m2/day q 28 days
Various
|
Nonmyeloid
|
Malignancies13 Cyclophosphamide
2.5 g/m2/day x 2
18
1/2
23 - 69 mcg/kg IV Etoposide
500 mg/m2/day x 3 days 8 - 28
Cisplatin
50 mg/m2/day x 3 q 28 days
Breast/Ovarian
|
Cancer14 Doxorubicin 75 mg/m2 21
2
11.5 mcg/kg IV
100 mg/m2 days 2 - 9
125 mg/m2 5.75 mcg/kg IV
150 mg/m2 days 10 - 12
q 14 days
Neuroblastoma
Cyclophosphamide
150 mg/m2 x 7 12
2
5.45 - 17.25 mcg/kg SC
Doxorubicin
35 mg/m2 days 6 - 19
75 mg/m2 Cisplatin
90 mg/m2 q 28 days
(cycles 1 3 5)
* NEU doses were those that accelerated neutrophil production. Doses which provided no additional acceleration beyond that achieved at the next lower dose are not reported.
Lowest dose(s) tested in the study.
Patients received doxorubicin at either 75 100 125 or 150 mg/m2.
Cycles 2 6 = cyclophosphamide 150 mg/m2 x 7 and etoposide 280 mg/m2 x 3.
Cycle 4 = cisplatin 90 mg/m2 x 1 and etoposide 280 mg/m2 x 3.
Patients With Acute Myeloid Leukemia Receiving Induction or Consolidation ChemotherapyIn a randomized, double-blind placebo-controlled multi-center phase 3 clinical trial 521 patients (median age 54 range 16 to 89 years) were treated for de novo acute myeloid leukemia (AML). Following a standard induction chemotherapy regimen comprising daunorubicin, cytosine arabinoside, and etoposide15 (DAV 3+7+5), patients received either NEUPOGEN at 5 mcg/kg/day or placebo, SC, from 24 hours after the last dose of chemotherapy until neutrophil recovery (ANC 1000/mm3 for 3 consecutive days or 10,000/mm3 for 1 day) or for a maximum of 35 days.
Treatment with NEUPOGEN significantly reduced the median time to ANC recovery and the median duration of fever, antibiotic use, and hospitalization following induction chemotherapy. In the NEUPOGEN -treated group the median time from initiation of chemotherapy to ANC recovery (ANC greater than or equal to 500/mm3) was 20 days (vs 25 days in the control group, p = 0.0001), the median duration of fever was reduced by 1.5 days (p = 0.009), and there were statistically significant reductions in the durations of IV antibiotic use and hospitalization. During consolidation therapy (DAV 2+5+5), patients treated with NEUPOGEN also experienced significant reductions in the incidence of severe neutropenia, time to neutrophil recovery, the incidence and duration of fever, and the durations of IV antibiotic use and hospitalization. Patients treated with a further course of standard (DAV 2+5+5) or high-dose cytosine arabinoside consolidation also experienced significant reductions in the duration of neutropenia.
There were no statistically significant differences between NEUPOGEN and placebo groups in complete remission rate (69% NEUPOGEN vs 68% placebo, p = 0.77), disease-free survival (median 342 days NEUPOGEN [n = 178], 322 days placebo [n = 177], p = 0.99), time to progression of all randomized patients (median 165 days NEUPOGEN , 186 days placebo, p = 0.87), or overall survival (median 380 days NEUPOGEN , 425 days placebo, p = 0.83).
Cancer Patients Receiving Bone Marrow TransplantIn two separate randomized controlled trials patients with Hodgkin s disease (HD) and non-Hodgkin s lymphoma (NHL) were treated with myeloablative chemotherapy and autologous bone marrow transplantation (ABMT). In one study (n = 54) NEUPOGEN was administered at doses of 10 or 30 mcg/kg/day; a third treatment group in this study received no NEUPOGEN . A statistically significant reduction in the median number of days of severe neutropenia (ANC less than 500/mm3) occurred in the NEUPOGEN -treated group versus the control group (23 days in the control group 11 days in the 10 mcg/kg/day group and 14 days in the 30 mcg/kg/day group [11 days in the combined treatment groups p = 0.004]). In the second study (n = 44 43 patients evaluable) NEUPOGEN was administered at doses of 10 or 20 mcg/kg/day; a third treatment group in this study received no NEUPOGEN . A statistically significant reduction in the median number of days of severe neutropenia occurred in the NEUPOGEN -treated group versus the control group (21.5 days in the control group and 10 days in both treatment groups p less than 0.001). The number of days of febrile neutropenia was also reduced significantly in this study (13.5 days in the control group 5 days in the 10 mcg/kg/day group and 5.5 days in the 20 mcg/kg/day group [5 days in the combined treatment groups p less than 0.0001]). Reductions in the number of days of hospitalization and antibiotic use were also seen although these reductions were not statistically significant. There were no effects on red blood cell or platelet levels.
In a randomized placebo-controlled trial 70 patients with myeloid and nonmyeloid malignancies were treated with myeloablative therapy and allogeneic bone marrow transplant followed by 300 mcg/m2/day of a Filgrastim product. A statistically significant reduction in the median number of days of severe neutropenia occurred in the treated group versus the control group (19 days in the control group and 15 days in the treatment group p less than 0.001) and time to recovery of ANC to greater than or equal to 500/mm3 (21 days in the control group and 16 days in the treatment group p less than 0.001).
In three nonrandomized studies (n = 119) patients received ABMT and treatment with NEUPOGEN . One study (n = 45) involved patients with breast cancer and malignant melanoma. A second study (n = 39) involved patients with HD. The third study (n = 35) involved patients with NHL acute lymphoblastic leukemia (ALL) and germ cell tumor. In these studies the recovery of the ANC to greater than or equal to 500/mm3 ranged from a median of 11.5 to 13 days.
None of the conditioning regimens used in the ABMT studies included radiation therapy.
While these studies were not designed to compare survival this information was collected and evaluated. The overall survival and disease progression of patients receiving NEUPOGEN in these studies were similar to those observed in the respective control groups and to historical data.
Peripheral Blood Progenitor Cell Collection and Therapy in Cancer PatientsAll patients in the Amgen-sponsored trials received a similar mobilization/collection regimen: NEUPOGEN was administered for 6 to 7 days with an apheresis procedure on days 5 6, and 7 (except for a limited number of patients receiving apheresis on days 4 6, and 8). In a non-Amgen-sponsored study patients underwent mobilization to a target number of mononuclear cells (MNC) with apheresis starting on day 5. There are no data on the mobilization of peripheral blood progenitor cells (PBPC) after days 4 to 5 that are not confounded by leukapheresis.
Mobilization: Mobilization of PBPC was studied in 50 heavily pretreated patients (median number of prior cycles = 9.5) with NHL HD, or ALL (Amgen study 1). CFU-GM was used as the marker for engraftable PBPC. The median CFU-GM level on each day of mobilization was determined from the data available (CFU-GM assays were not obtained on all patients on each day of mobilization). These data are presented below.
The data from Amgen study 1 were supported by data from Amgen study 2 in which 22 pretreated breast cancer patients (median number of prior cycles = 3) were studied. Both the CFU-GM and CD34+ cells reached a maximum on day 5 at greater than 10-fold over baseline and then remained elevated with leukapheresis.
* n/a = not available
Progenitor Cell Levels in Peripheral Blood by Mobilization Day No.
SamplesMedian
(25% - 75%)No.
SamplesMedian
(25% - 75%)No.
SamplesMedian
(25% - 75%)Day 1
11
18
(13 - 62)
20
42
(15 - 151)
20
0.13
(0.02 - 0.66)
Day 2
7
22
(3 - 61)
n/a*
n/a*
n/a*
n/a*
Day 3
10
138
(39 - 364)
n/a*
n/a*
n/a*
n/a*
Day 4
18
365
(158 - 864)
18
576
(108 - 1819)
17
2.11
(0.58 - 3.93)
Day 5
36
781
(391 - 1608)
21
960
(72 - 1677)
22
3.16
(1.08 - 6.11)
Day 6
46
505
(199 - 1397)
22
756
(70 - 3486)
22
2.67
(1.09 - 4.40)
Day 7
37
333
(111 - 938)
22
597
(118 - 2009)
21
2.64
(0.78 - 4.22)
Day 8
15
383
(94 - 815)
12
51
(10 -746)
12
1.61
(0.38 - 4.31)
In three studies of patients with prior exposure to chemotherapy the median CFU-GM yield in the leukapheresis product ranged from 20.9 to 32.7 x 104/kg body weight (n = 105). In two of these studies where CD34+ yields in the leukapheresis product were also determined the median CD34+ yields were 3.11 and 2.80 x 106/kg, respectively (n = 56). In an additional study of 18 chemotherapy-naive patients the median CFU-GM yield was 123.4 x 104/kg.
Engraftment: Engraftment following NEUPOGEN -mobilized PBPC is summarized for 101 patients in the following table. In all studies, a Cox regression model showed that the total number of CFU-GM and/or CD34+ cells collected was a significant predictor of time to platelet recovery.
In a randomized, unblinded study of patients with HD or NHL undergoing myeloablative chemotherapy (Amgen study 3) 27 patients received NEUPOGEN -mobilized PBPC followed by NEUPOGEN and 31 patients received ABMT followed by NEUPOGEN . Patients randomized to the NEUPOGEN -mobilized PBPC group compared to the ABMT group had significantly fewer days of platelet transfusions (median 6 vs 10 days) a significantly shorter time to a sustained platelet count greater than 20 000/mm3 (median 16 vs 23 days) a significantly shorter time to recovery of a sustained ANC greater than or equal to 500/mm3 (median 11 vs 14 days) significantly fewer days of red blood cell transfusions (median 2 vs 3 days) and a significantly shorter duration of posttransplant hospitalization.
* n/a = not available
Amgen-sponsored
Study 1
N = 13Amgen-sponsored
Study 2
N = 22Amgen-sponsored
Study 3
N = 27Non-Amgen-
sponsored Study
N = 39Median PBPC/kg Collected
MNC
9.5 x 108 9.5 x 108 8.1 x 108 10.3 x 108 CD34+ n/a* 3.1 x 106 2.8 x 106 6.2 x 106 CFU-GM
63.9 x 104 25.3 x 104 32.6 x 104 n/a* Days to ANC 500/mm3
Median
9
10
11
10
Range
8 - 10
8 - 15
9 - 38
7 - 40 Days to Plt. 20,00/mm3
Median
10
12.5
16
15.5
Range
7 - 16
10 - 30
8 - 52
7 - 63
Three of the 101 patients (3%) did not achieve the criteria for engraftment as defined by a platelet count greater than or equal to 20 000/mm3 by day 28. In clinical trials of NEUPOGEN for the mobilization of PBPC NEUPOGEN was administered to patients at 5 to 24 mcg/kg/day after reinfusion of the collected cells until a sustainable ANC (greater than or equal to 500/mm3) was reached. The rate of engraftment of these cells in the absence of NEUPOGEN posttransplantation has not been studied.
Patients With Severe Chronic NeutropeniaSevere chronic neutropenia (SCN) (idiopathic cyclic and congenital) is characterized by a selective decrease in the number of circulating neutrophils and an enhanced susceptibility to bacterial infections.
The daily administration of NEUPOGEN has been shown to be safe and effective in causing a sustained increase in the neutrophil count and a decrease in infectious morbidity in children and adults with the clinical syndrome of SCN.16 In the phase 3 trial summarized in the following table daily treatment with NEUPOGEN resulted in significant beneficial changes in the incidence and duration of infection fever antibiotic use and oropharyngeal ulcers. In this trial 120 patients with a median age of 12 years (range 1 to 76 years) were treated.
* Incidence values were calculated for each patient and are defined as the total number of events experienced divided by the number of 28-day periods of exposure (on-study). Median incidence values were then reported for each patient group.
Control patients were observed for a 4-month period.The incidence for each of these 5 clinical parameters was lower in the NEUPOGEN arm compared to the control arm for cohorts in each of the 3 major diagnostic categories. All 3 diagnostic groups showed favorable trends in favor of treatment. An analysis of variance showed no significant interaction between treatment and diagnosis suggesting that efficacy did not differ substantially in the different diseases. Although NEUPOGEN substantially reduced neutropenia in all patient groups in patients with cyclic neutropenia cycling persisted but the period of neutropenia was shortened to 1 day.
As a result of the lower incidence and duration of infections there was also a lower number of episodes of hospitalization (28 hospitalizations in 62 patients in the treated group vs 44 hospitalizations in 60 patients in the control group over a 4-month period [p = 0.0034]). Patients treated with NEUPOGEN also reported a lower number of episodes of diarrhea nausea fatigue and sore throat.
In the phase 3 trial untreated patients had a median ANC of 210/mm3 (range 0 to 1550/mm3). NEUPOGEN therapy was adjusted to maintain the median ANC between 1500 and 10 000/mm3. Overall the response to NEUPOGEN was observed in 1 to 2 weeks. The median ANC after 5 months of NEUPOGEN therapy for all patients was 7460/mm3 (range 30 to 30 880/mm3). NEUPOGEN dosing requirements were generally higher for patients with congenital neutropenia (2.3 to 40 mcg/kg/day) than for patients with idiopathic (0.6 to 11.5 mcg/kg/day) or cyclic (0.5 to 6 mcg/kg/day) neutropenia.
Warnings ⮝
Allergic ReactionsAllergic-type reactions occurring on initial or subsequent treatment have been reported in less than 1 in 4000 patients treated with NEUPOGEN . These have generally been characterized by systemic symptoms involving at least 2 body systems most often skin (rash urticaria facial edema) respiratory (wheezing dyspnea) and cardiovascular (hypotension tachycardia). Some reactions occurred on initial exposure. Reactions tended to occur within the first 30 minutes after administration and appeared to occur more frequently in patients receiving NEUPOGEN IV. Rapid resolution of symptoms occurred in most cases after administration of antihistamines steroids bronchodilators and/or epinephrine. Symptoms recurred in more than half the patients who were rechallenged.
SPLENIC RUPTURESPLENIC RUPTURE, INCLUDING FATAL CASES, HAS BEEN REPORTED FOLLOWING THE ADMINISTRATION OF NEUPOGEN . INDIVIDUALS RECEIVING NEUPOGEN WHO REPORT LEFT UPPER ABDOMINAL AND/OR SHOULDER TIP PAIN SHOULD BE EVALUATED FOR AN ENLARGED SPLEEN OR SPLENIC RUPTURE.
Acute Respiratory Distress Syndrome (ARDS)Acute respiratory distress syndrome (ARDS) has been reported in patients receiving NEUPOGEN , and is postulated to be secondary to an influx of neutrophils to sites of inflammation in the lungs. Patients receiving NEUPOGEN who develop fever, lung infiltrates, or respiratory distress should be evaluated for the possibility of ARDS. In the event that ARDS occurs, NEUPOGEN should be withheld until resolution of ARDS or discontinued. Patients should receive appropriate medical management for this condition.
Alveolar Hemorrhage and HemoptysisAlveolar hemorrhage manifesting as pulmonary infiltrates and hemoptysis requiring hospitalization has been reported in healthy donors undergoing peripheral blood progenitor cell (PBPC) mobilization. Hemoptysis resolved with discontinuation of NEUPOGEN . The use of NEUPOGEN for PBPC mobilization in healthy donors is not an approved indication.
Sickle Cell DisordersSevere sickle cell crises, in some cases resulting in death, have been associated with the use of NEUPOGEN in patients with sickle cell disorders. Only physicians qualified by specialized training or experience in the treatment of patients with sickle cell disorders should prescribe NEUPOGEN for such patients, and only after careful consideration of the potential risks and benefits.
Patients With Severe Chronic NeutropeniaThe safety and efficacy of NEUPOGEN in the treatment of neutropenia due to other hematopoietic disorders (eg myelodysplastic syndrome [MDS]) have not been established. Care should be taken to confirm the diagnosis of SCN before initiating NEUPOGEN therapy.
MDS and AML have been reported to occur in the natural history of congenital neutropenia without cytokine therapy.17 Cytogenetic abnormalities, transformation to MDS, and AML have also been observed in patients treated with NEUPOGEN for SCN. Based on available data including a postmarketing surveillance study, the risk of developing MDS and AML appears to be confined to the subset of patients with congenital neutropenia (see ADVERSE REACTIONS). Abnormal cytogenetics and MDS have been associated with the eventual development of myeloid leukemia. The effect of NEUPOGEN on the development of abnormal cytogenetics and the effect of continued NEUPOGEN administration in patients with abnormal cytogenetics or MDS are unknown. If a patient with SCN develops abnormal cytogenetics or myelodysplasia the risks and benefits of continuing NEUPOGEN should be carefully considered.
Precautions ⮝
GeneralSimultaneous Use With Chemotherapy and Radiation TherapyThe safety and efficacy of NEUPOGEN given simultaneously with cytotoxic chemotherapy have not been established. Because of the potential sensitivity of rapidly dividing myeloid cells to cytotoxic chemotherapy do not use NEUPOGEN in the period 24 hours before through 24 hours after the administration of cytotoxic chemotherapy (see DOSAGE AND ADMINISTRATION).
The efficacy of NEUPOGEN has not been evaluated in patients receiving chemotherapy associated with delayed myelosuppression (eg, nitrosoureas) or with mitomycin C or with myelosuppressive doses of antimetabolites such as 5-fluorouracil.
The safety and efficacy of NEUPOGEN have not been evaluated in patients receiving concurrent radiation therapy. Simultaneous use of NEUPOGEN with chemotherapy and radiation therapy should be avoided.
Potential Effect on Malignant CellsNEUPOGEN is a growth factor that primarily stimulates neutrophils. However the possibility that NEUPOGEN can act as a growth factor for any tumor type cannot be excluded. In a randomized study evaluating the effects of NEUPOGEN versus placebo in patients undergoing remission induction for AML, there was no significant difference in remission rate, disease-free, or overall survival (see CLINICAL EXPERIENCE).
The safety of NEUPOGEN in chronic myeloid leukemia (CML) and myelodysplasia has not been established.
When NEUPOGEN is used to mobilize PBPC tumor cells may be released from the marrow and subsequently collected in the leukapheresis product. The effect of reinfusion of tumor cells has not been well studied and the limited data available are inconclusive.
LeukocytosisCancer Patients Receiving Myelosuppressive ChemotherapyWhite blood cell counts of 100 000/mm3 or greater were observed in approximately 2% of patients receiving NEUPOGEN at doses above 5 mcg/kg/day. There were no reports of adverse events associated with this degree of leukocytosis. In order to avoid the potential complications of excessive leukocytosis a CBC is recommended twice per week during NEUPOGEN therapy (see LABORATORY MONITORING).
Premature Discontinuation of NEUPOGEN TherapyCancer Patients Receiving Myelosuppressive ChemotherapyA transient increase in neutrophil counts is typically seen 1 to 2 days after initiation of NEUPOGEN therapy. However for a sustained therapeutic response NEUPOGEN therapy should be continued following chemotherapy until the post nadir ANC reaches 10 000/mm3. Therefore the premature discontinuation of NEUPOGEN therapy prior to the time of recovery from the expected neutrophil nadir is generally not recommended (see DOSAGE AND ADMINISTRATION).
ImmunogenicityAs with all therapeutic proteins, there is a potential for immunogenicity. The incidence of antibody development in patients receiving NEUPOGEN has not been adequately determined. While available data suggest that a small proportion of patients developed binding antibodies to Filgrastim, the nature and specificity of these antibodies has not been adequately studied. In clinical studies comparing NEUPOGEN and Neulasta , the incidence of antibodies binding to NEUPOGEN was 3% (11/333). In these 11 patients, no evidence of a neutralizing response was observed using a cell-based bioassay. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay, and the observed incidence of antibody positivity in an assay may be influenced by several factors including timing of sampling, sample handling, concomitant medications, and underlying disease. Therefore, comparison of the incidence of antibodies to NEUPOGEN with the incidence of antibodies to other products may be misleading.
Cytopenias resulting from an antibody response to exogenous growth factors have been reported on rare occasions in patients treated with other recombinant growth factors. There is a theoretical possibility that an antibody directed against Filgrastim may cross-react with endogenous G-CSF, resulting in immune-mediated neutropenia; however, this has not been reported in clinical studies or in post-marketing experience. Patients who develop hypersensitivity to Filgrastim (NEUPOGEN ) may have allergic or hypersensitivity reactions to other E coli-derived proteins.
Cutaneous VasculitisCutaneous vasculitis has been reported in patients treated with NEUPOGEN . In most cases the severity of cutaneous vasculitis was moderate or severe. Most of the reports involved patients with SCN receiving long-term NEUPOGEN therapy. Symptoms of vasculitis generally developed simultaneously with an increase in the ANC and abated when the ANC decreased. Many patients were able to continue NEUPOGEN at a reduced dose.
Information for Patients and CaregiversPatients should be referred to the Information for Patients and Caregivers labeling included with the package insert in each dispensing pack of NEUPOGEN vials or NEUPOGEN prefilled syringes. The Information for Patients and Caregivers labeling provides information about neutrophils and neutropenia and the safety and efficacy of NEUPOGEN . It is not intended to be a disclosure of all known or possible effects.
Laboratory MonitoringCancer Patients Receiving Myelosuppressive ChemotherapyA CBC and platelet count should be obtained prior to chemotherapy and at regular intervals (twice per week) during NEUPOGEN therapy. Following cytotoxic chemotherapy the neutrophil nadir occurred earlier during cycles when NEUPOGEN was administered and WBC differentials demonstrated a left shift including the appearance of promyelocytes and myeloblasts. In addition the duration of severe neutropenia was reduced and was followed by an accelerated recovery in the neutrophil counts.
Cancer Patients Receiving Bone Marrow TransplantFrequent CBCs and platelet counts are recommended (at least 3 times per week) following marrow transplantation.
Patients With Severe Chronic NeutropeniaDuring the initial 4 weeks of NEUPOGEN therapy and during the 2 weeks following any dose adjustment a CBC with differential and platelet count should be performed twice weekly. Once a patient is clinically stable a CBC with differential and platelet count should be performed monthly during the first year of treatment. Thereafter, if clinically stable, routine monitoring with regular CBCs (ie, as clinically indicated but at least quarterly) is recommended. Additionally, for those patients with congenital neutropenia, annual bone marrow and cytogenetic evaluations should be performed throughout the duration of treatment (see WARNINGS, ADVERSE REACTIONS).
In clinical trials the following laboratory results were observed:
Drug Interaction
- Cyclic fluctuations in the neutrophil counts were frequently observed in patients with congenital or idiopathic neutropenia after initiation of NEUPOGEN therapy.
- Platelet counts were generally at the upper limits of normal prior to NEUPOGEN therapy. With NEUPOGEN therapy platelet counts decreased but usually remained within normal limits (see ADVERSE REACTIONS).
- Early myeloid forms were noted in peripheral blood in most patients including the appearance of metamyelocytes and myelocytes. Promyelocytes and myeloblasts were noted in some patients.
- Relative increases were occasionally noted in the number of circulating eosinophils and basophils. No consistent increases were observed with NEUPOGEN therapy.
- As in other trials increases were observed in serum uric acid lactic dehydrogenase and serum alkaline phosphatase.
Drug interactions between NEUPOGEN and other drugs have not been fully evaluated. Drugs which may potentiate the release of neutrophils such as lithium should be used with caution.
Increased hematopoietic activity of the bone marrow in response to growth factor therapy has been associated with transient positive bone imaging changes. This should be considered when interpreting bone-imaging results.
Carcinogenesis, Mutagenesis, Impairment of FertilityThe carcinogenic potential of NEUPOGEN has not been studied. NEUPOGEN failed to induce bacterial gene mutations in either the presence or absence of a drug metabolizing enzyme system. NEUPOGEN had no observed effect on the fertility of male or female rats or on gestation at doses up to 500 mcg/kg.
Pregnancy Category CNEUPOGEN has been shown to have adverse effects in pregnant rabbits when given in doses 2 to 10 times the human dose. Since there are no adequate and well-controlled studies in pregnant women, the effect, if any, of NEUPOGEN on the developing fetus or the reproductive capacity of the mother is unknown. However, the scientific literature describes transplacental passage of NEUPOGEN when administered to pregnant rats during the latter part of gestation18 and apparent transplacental passage of NEUPOGEN when administered to pregnant humans by 30 hours prior to preterm delivery ( 30 weeks gestation).19 NEUPOGEN should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
In rabbits increased abortion and embryolethality were observed in animals treated with NEUPOGEN at 80 mcg/kg/day. NEUPOGEN administered to pregnant rabbits at doses of 80 mcg/kg/day during the period of organogenesis was associated with increased fetal resorption genitourinary bleeding developmental abnormalities decreased body weight live births and food consumption. External abnormalities were not observed in the fetuses of dams treated at 80 mcg/kg/day. Reproductive studies in pregnant rats have shown that NEUPOGEN was not associated with lethal teratogenic or behavioral effects on fetuses when administered by daily IV injection during the period of organogenesis at dose levels up to 575 mcg/kg/day.
In Segment III studies in rats offspring of dams treated at > 20 mcg/kg/day exhibited a delay in external differentiation (detachment of auricles and descent of testes) and slight growth retardation possibly due to lower body weight of females during rearing and nursing. Offspring of dams treated at 100 mcg/kg/day exhibited decreased body weights at birth and a slightly reduced 4-day survival rate.
Nursing MothersIt is not known whether NEUPOGEN is excreted in human milk. Because many drugs are excreted in human milk caution should be exercised if NEUPOGEN is administered to a nursing woman.
Pediatric UseIn a phase 3 study to assess the safety and efficacy of NEUPOGEN in the treatment of SCN, 120 patients with a median age of 12 years were studied. Of the 120 patients, 12 were infants (1 month to 2 years of age), 47 were children (2 to 12 years of age), and 9 were adolescents (12 to 16 years of age). Additional information is available from a SCN postmarketing surveillance study, which includes long-term follow-up of patients in the clinical studies and information from additional patients who entered directly into the postmarketing surveillance study. Of the 531 patients in the surveillance study as of 31 December 1997, 32 were infants, 200 were children, and 68 were adolescents (see CLINICAL EXPERIENCE, INDICATIONS AND USAGE, LABORATORY MONITORING, DOSAGE AND ADMINISTRATION).
Pediatric patients with congenital types of neutropenia (Kostmann s syndrome, congenital agranulocytosis, or Schwachman-Diamond syndrome) have developed cytogenetic abnormalities and have undergone transformation to MDS and AML while receiving chronic NEUPOGEN treatment. The relationship of these events to NEUPOGEN administration is unknown (see WARNINGS, ADVERSE REACTIONS).
Long-term follow-up data from the postmarketing surveillance study suggest that height and weight are not adversely affected in patients who received up to 5 years of NEUPOGEN treatment. Limited data from patients who were followed in the phase 3 study for 1.5 years did not suggest alterations in sexual maturation or endocrine function.
The safety and efficacy in neonates and patients with autoimmune neutropenia of infancy have not been established.
In the cancer setting 12 pediatric patients with neuroblastoma have received up to 6 cycles of cyclophosphamide cisplatin doxorubicin and etoposide chemotherapy concurrently with NEUPOGEN ; in this population NEUPOGEN was well tolerated. There was one report of palpable splenomegaly associated with NEUPOGEN therapy; however the only consistently reported adverse event was musculoskeletal pain which is no different from the experience in the adult population.
Geriatric UseAmong 855 subjects enrolled in 3 randomized, placebo-controlled trials of NEUPOGEN use following myelosuppressive chemotherapy, there were 232 subjects age 65 or older, and 22 subjects age 75 or older. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other clinical experience has not identified differences in the responses between elderly and younger patients.
Clinical studies of NEUPOGEN in other approved indications (ie, bone marrow transplant recipients, PBPC mobilization, and SCN) did not include sufficient numbers of subjects aged 65 and older to determine whether elderly subjects respond differently from younger subjects.
Overdosage ⮝
In cancer patients receiving NEUPOGEN as an adjunct to myelosuppressive chemotherapy it is recommended to avoid the potential risks of excessive leukocytosis that NEUPOGEN therapy be discontinued if the ANC surpasses 10 000/mm3 after the chemotherapy-induced ANC nadir has occurred. Doses of NEUPOGEN that increase the ANC beyond 10 000/mm3 may not result in any additional clinical benefit.
The maximum tolerated dose of NEUPOGEN has not been determined. Efficacy was demonstrated at doses of 4 to 8 mcg/kg/day in the phase 3 study of nonmyeloablative chemotherapy. Patients in the BMT studies received up to 138 mcg/kg/day without toxic effects although there was a flattening of the dose response curve above daily doses of greater than 10 mcg/kg/day.
In NEUPOGEN clinical trials of cancer patients receiving myelosuppressive chemotherapy WBC counts > 100 000/mm3 have been reported in less than 5% of patients but were not associated with any reported adverse clinical effects.
In cancer patients receiving myelosuppressive chemotherapy discontinuation of NEUPOGEN therapy usually results in a 50% decrease in circulating neutrophils within 1 to 2 days with a return to pretreatment levels in 1 to 7 days.
How Supplied ⮝
NEUPOGEN : Use only one dose per vial; do not re-enter the vial. Discard unused portions. Do not save unused drug for later administration.
Use only one dose per prefilled syringe. Discard unused portions. Do not save unused drug for later administration.
VialsSingle-dose preservative-free vials containing 300 mcg (1 mL) of Filgrastim (300 mcg/mL). 1 Vial (NDC 54868-2522-0); Dispensing packs of 10 (NDC 54868-2522-1).
Prefilled Syringes (SingleJect )
Single-dose preservative-free, prefilled syringes with 27 gauge, inch needles with an UltraSafe Needle Guard, containing 300 mcg (0.5 mL) of Filgrastim (600 mcg/mL). Dispensing packs of 10 (NDC 54868-5020-0).
Single-dose preservative-free, prefilled syringes with 27 gauge, inch needles with an UltraSafe Needle Guard, containing 480 mcg (0.8 mL) of Filgrastim (600 mcg/mL). Dispensing packs of 10 (NDC 54868-3050-0).
The needle cover of the prefilled syringe contains dry natural rubber (a derivative of latex).
NEUPOGEN should be stored at 2 to 8 C (36 to 46 F). Avoid shaking.
References ⮝
- Zsebo KM Cohen AM Murdock DC et al. Recombinant human granulocyte colony-stimulating factor: Molecular and biological characterization. Immunobiol. 1986;172:175-184.
- Welte K Bonilla MA Gillio AP et al. Recombinant human G-CSF: Effects on hematopoiesis in normal and cyclophosphamide treated primates. J Exp Med. 1987;165:941-948.
- Duhrsen U Villeval JL Boyd J et al. Effects of recombinant human granulocyte colony-stimulating factor on hematopoietic progenitor cells in cancer patients. Blood. 1988;72:2074-2081.
- Souza LM Boone TC Gabrilove J et al. Recombinant human granulocyte colony-stimulating factor: Effects on normal and leukemic myeloid cells. Science. 1986;232:61-65.
- Weisbart RH Kacena A Schuh A Golde DW. GM-CSF induces human neutrophil IgA-mediated phagocytosis by an IgA Fc receptor activation mechanism. Nature. 1988;332:647-648.
- Kitagawa S Yuo A Souza LM Saito M Miura Y Takaku F. Recombinant human granulocyte colony-stimulating factor enhances superoxide release in human granulocytes stimulated by chemotactic peptide. Biochem Biophys Res Commun. 1987;1443:1146.
- Glaspy JA Baldwin GC Robertson PA et al. Therapy for neutropenia in hairy cell leukemia with recombinant human granulocyte colony-stimulating factor. Ann Int Med. 1988;109:789-795.
- Yuo A Kitagawa S Ohsaka A et al. Recombinant human granulocyte colony-stimulating factor as an activator of human granulocytes: Potentiation of responses triggered by receptor-mediated agonists and stimulation of C3bi receptor expression and adherence. Blood. 1989;74:2144-2149.
- Gabrilove JL Jakubowski A Fain K et al. Phase I study of granulocyte colony-stimulating factor in patients with transitional cell carcinoma of the urothelium. J Clin Invest. 1988;82:1454-1461.
- Morstyn G Souza L Keech J et al. Effect of granulocyte colony-stimulating factor on neutropenia induced by cytotoxic chemotherapy. Lancet. 1988;1:667-672.
- Bronchud MH Scarffe JH Thatcher N et al. Phase I/II study of recombinant human granulocyte colony-stimulating factor in patients receiving intensive chemotherapy for small cell lung cancer. Br J Cancer. 1987;56:809-813.
- Gabrilove JL Jakubowski A Scher H et al. Effect of granulocyte colony-stimulating factor on neutropenia and associated morbidity due to chemotherapy for transitional cell carcinoma of the urothelium. N Engl J Med. 1988;318:1414-1422.
- Neidhart J Mangalik A Kohler W et al. Granulocyte colony-stimulating factor stimulates recovery of granulocytes in patients receiving dose-intensive chemotherapy without bone-marrow transplantation. J Clin Oncol. 1989;7:1685-1691.
- Bronchud MH Howell A Crowther D et al. The use of granulocyte colony-stimulating factor to increase the intensity of treatment with doxorubicin in patients with advanced breast and ovarian cancer. Br J Cancer. 1989;60:121-128.
- Heil G, Hoelzer D, Sanz MA, et al. A randomized, double-blind, placebo-controlled, phase III study of Filgrastim in remission induction and consolidation therapy for adults with de novo Acute Myeloid Leukemia. Blood. 1997;90:4710-4718.
- Dale DC Bonilla MA Davis MW et al. A randomized controlled phase III trial of recombinant human granulocyte colony-stimulating factor (Filgrastim) for treatment of severe chronic neutropenia. Blood. 1993;81:2496-2502.
- Schroeder TM and Kurth R. Spontaneous chromosomal breakage and high incidence of leukemia in inherited disease. Blood. 1971;37:96-112.
- Medlock ES, Kaplan DL, Cecchini M, Ulich TR, del Castillo J, Andresen J. Granulocyte colony-stimulating factor crosses the placenta and stimulates fetal rat granulopoiesis. Blood. 1993;81:916-922.
- Calhoun DA, Rosa C, Christensen RD. Transplacental passage of recombinant human granulocyte colony-stimulating factor in women with an imminent preterm delivery. Am J Obstet Gynecol. 1996;174:1306-1311.
This product and its use are covered by the following US Patent Nos.: 4,810,643; 4,999,291; 5,582,823; 5,580,755.
[AMGEN LOGO]
Manufactured by:
Amgen Manufacturing, Limited,
a subsidiary of Amgen Inc.
One Amgen Center Drive
Thousand Oaks, California 91320-17993xxxxxx
1991-2007 Amgen Inc. All rights reserved.
v.20.2 - Issue Date: 09/2007
NEUPOGEN
(Filgrastim)
Information for Patients and CaregiversThis patient package insert provides information and instructions for people who will be receiving NEUPOGEN and their caregivers. This patient package insert does not tell you everything about NEUPOGEN . You should discuss any questions you have about treatment with NEUPOGEN with your doctor.
What is NEUPOGEN ?
NEUPOGEN is a man-made form of granulocyte colony-stimulating factor (G-CSF), which is made using the bacteria E coli. G-CSF is a substance naturally produced by the body. It stimulates the growth of neutrophils (nu-tro-fils), a type of white blood cell important in the body s fight against infection.
What is NEUPOGEN used for?
NEUPOGEN is used to treat neutropenia (nu-tro-peen-ee-ah), a condition where the body makes too few neutrophils. Neutropenia may be a long-standing condition where your body does not make enough neutrophils, or it may be caused by drugs used to treat cancer. In some cases, your body may make enough neutrophils, but as part of your treatment for cancer, your doctor may want to increase the number of certain blood cells (CD34 cells) and collect them. The cells are collected using a process called apheresis (ay-fer-ree-sis). These collected cells are given back to you after you receive very high doses of treatment for cancer to make your blood counts get back to normal more quickly.
How does NEUPOGEN work?
NEUPOGEN works by helping your body make more neutrophils. To make sure NEUPOGEN is working, your doctor will ask that you have regular blood tests to count the number of neutrophils you have. It is important that you follow your doctor s instructions about getting these tests.
Who should not take NEUPOGEN ?
Do not take NEUPOGEN if you are:
- Allergic to NEUPOGEN (Filgrastim) or any of its ingredients. See the end of this leaflet for a list of ingredients in NEUPOGEN .
- Allergic to other medicines made using the bacteria E coli. Ask your doctor if you are not sure.
What important information do I need to know about taking NEUPOGEN ?
NEUPOGEN may reduce your chance of getting an infection, but does not prevent all infections. An infection can still happen during the short time when your/your child's neutrophil levels are low. You must be alert and look for some of the common signs or symptoms of infection, such as fever, chills, rash, sore throat, diarrhea, or redness, swelling, or pain around a cut or sore. If you/your child has any of these signs or symptoms during treatment with NEUPOGEN , tell your doctor or nurse immediately.
There is a possibility that you could have a reaction at an injection site. If there is a lump, swelling, or bruising at an injection site that does not go away, call your doctor.
If you have a sickle cell disorder, make sure that you tell your doctor before you start taking NEUPOGEN . If you have a sickle cell crisis after getting NEUPOGEN , tell your doctor right away.
Make sure your doctor knows about all medicines, and herbal or vitamin supplements you are taking before starting NEUPOGEN . If you are taking lithium you may need more frequent blood tests.
If you/your child are receiving NEUPOGEN because you are also receiving chemotherapy, the last dose of NEUPOGEN should be injected at least 24 hours before your next dose of chemotherapy.
There is more information about NEUPOGEN in the Physician Package Insert. If you have any questions, you should talk to your doctor.
What are possible serious side effects of NEUPOGEN ?
- Spleen Rupture. Your spleen may become enlarged and can rupture while taking NEUPOGEN . A ruptured spleen can cause death. The spleen is located in the upper left section of your stomach area. Call your doctor right away if you or your child has pain in the left upper stomach area or left shoulder tip area. This pain could mean your or your child s spleen is enlarged or ruptured.
- Serious Allergic Reactions. NEUPOGEN can cause serious allergic reactions. These reactions can cause a rash over the whole body, shortness of breath, wheezing, dizziness, swelling around the mouth or eyes, fast pulse, and sweating. If you or your child starts to have any of these symptoms, stop using NEUPOGEN and call your doctor or seek emergency care right away. If you or your child has an allergic reaction during the injection of NEUPOGEN , stop the injection right away.
- A serious lung problem called acute respiratory distress syndrome (ARDS). Call your doctor or seek emergency care right away if you or your child has shortness of breath, trouble breathing or a fast rate of breathing.
What are the most common side effects of NEUPOGEN ?
The most common side effect you/your child may experience is aching in the bones and muscles. This aching can usually be relieved by taking a non-aspirin pain reliever such as acetaminophen.
Some people experience redness, swelling, or itching at the site of injection. This may be an allergy to the ingredients in NEUPOGEN , or it may be a local reaction. If you are giving an injection to a child, look for signs of redness, swelling, or itching at the site of injection because they may not be able to tell you they are experiencing a reaction. If you notice any signs of a local reaction, call your doctor.
What about pregnancy or breastfeeding?
NEUPOGEN has not been studied in pregnant women, and its effects on unborn babies are not known. If you take NEUPOGEN while you are pregnant, it is possible that small amounts of it may get into your baby s blood. It is not known if NEUPOGEN can get into human breast milk.
If you are pregnant, plan to become pregnant, think you may be pregnant, or are breast feeding, you should tell your doctor before using NEUPOGEN .
How to prepare and give a NEUPOGEN injection
NEUPOGEN should be injected at the same time each day. If you miss a dose contact your doctor or nurse.
You must always use the correct dose of NEUPOGEN . Too little NEUPOGEN may not protect you against infections, and too much NEUPOGEN may cause too many neutrophils to be in your blood. Your doctor will determine your/your child s correct dose based on your/your child's body weight.
If you are giving someone else NEUPOGEN injections, it is important that you know how to inject NEUPOGEN , how much to inject, and how often to inject NEUPOGEN .
NEUPOGEN is available as a liquid in vials or in prefilled syringes. When you receive your NEUPOGEN , always check to see that:
- The name NEUPOGEN appears on the package and vial or prefilled syringe label.
- The expiration date on the vial or prefilled syringe label has not passed. You should not use a vial or prefilled syringe after the date on the label.
- The strength of the NEUPOGEN (number of micrograms in the colored dot on the package containing the vial or prefilled syringe) is the same as your doctor prescribed.
- The NEUPOGEN liquid in the vial or in the prefilled syringe is clear and colorless. Do not use NEUPOGEN if the contents of the vial or prefilled syringe appear discolored or cloudy, or if the vial or prefilled syringe appears to contain lumps, flakes, or particles.
only use the syringe that your doctor prescribes.
Your doctor or nurse will give you instructions on how to measure the correct dose of NEUPOGEN . This dose will be measured in milliliters. You should only use a syringe that is marked in tenths of milliliters, or mL (for example, 0.2 mL). The doctor or nurse may refer to an mL as a cc (1 mL = 1 cc). If you do not use the correct syringe, you or your child could receive too much or too little NEUPOGEN .
Only use disposable syringes and needles. Use the syringes only once and dispose of them as instructed by your doctor or nurse.
IMPORTANT: TO HELP AVOID POSSIBLE INFECTION, YOU SHOULD FOLLOW THESE INSTRUCTIONS.
Setting up for an injection
- Find a clean flat working surface, such as a table.
- Remove the vial or prefilled syringe of NEUPOGEN from the refrigerator. Allow NEUPOGEN to reach room temperature (this takes about 30 minutes). Vials or prefilled syringes should be used only once. DO NOT SHAKE THE VIAL OR PREFILLED SYRINGE. Shaking may damage the NEUPOGEN . If the vial or prefilled syringe has been shaken vigorously, the solution may appear foamy and it should not be used.
- Assemble the supplies you will need for an injection:
- NEUPOGEN vial and disposable syringe and needle


- NEUPOGEN prefilled syringe with transparent (clear) plastic orange needle guard attached

- two alcohol swabs and one cotton ball or gauze pad

- puncture-proof disposal container
4. Wash your hands with soap and warm water.

HOW TO PREPARE THE DOSE OF NEUPOGEN IN VIALS OR PREFILLED SYRINGES
If you are using NEUPOGEN in a vial, follow the instructions in Section A. If you are using NEUPOGEN in a prefilled syringe, go to Section B.
Section A. Preparing the dose of NEUPOGEN in a vial
1. Take the cap off the vial. Clean the rubber stopper with one alcohol swab. 2. Check the package containing the syringe. If the package has been opened or damaged, do not use that syringe. Dispose of that syringe in the puncture-proof disposal container. If the syringe package is undamaged, open the package and remove the syringe. 3. Pull the needle cover straight off the syringe. Then, pull back the plunger and draw air into the syringe. The amount of air drawn into the syringe should be the same amount (mL or cc) as the dose of NEUPOGEN that your doctor prescribed. 4. Keep the vial on your flat working surface and insert the needle straight down through the rubber stopper. Do not put the needle through the rubber stopper more than once. 5. Push the plunger of the syringe down and inject the air from the syringe into the vial of NEUPOGEN . 6. Keeping the needle in the vial, turn the vial upside down. Make sure that the NEUPOGEN liquid is covering the tip of the needle. 7. Keeping the vial upside down, slowly pull back on the plunger to fill the syringe with NEUPOGEN liquid to the number (mL or cc) that matches the dose your doctor prescribed. 8. Keeping the needle in the vial, check for air bubbles in the syringe. If there are air bubbles, gently tap the syringe with your fingers until the air bubbles rise to the top of the syringe. Then slowly push the plunger up to force the air bubbles out of the syringe. 9. Keeping the tip of the needle in the liquid, once again pull the plunger back to the number on the syringe that matches your dose. Check again for air bubbles. The air in the syringe will not hurt you, but too large an air bubble can reduce your dose of NEUPOGEN . If there are still air bubbles, repeat the steps above to remove them. 10. Check again to make sure that you have the correct dose in the syringe. It is important that you use the exact dose prescribed by your doctor. Remove the syringe from the vial but do not lay it down or let the needle touch anything. (Go to Injecting the dose of NEUPOGEN ).
Section B. Preparing the dose of NEUPOGEN in a prefilled syringe
- Remove the syringe from the package and the tray. Check to see that the plastic orange needle guard is covering the barrel of the glass syringe. DO NOT push the orange needle guard over the needle cover before injection. This may activate or lock the needle guard. If the orange needle guard is covering the needle that means it has been activated. Do NOT use that syringe. Dispose of that syringe in the puncture-proof disposal container. Use a new syringe from the package.
- Hold the syringe barrel through the needle guard windows with the needle pointing up. Holding the syringe with the needle pointing up helps to prevent medicine from leaking out of the needle. Carefully pull the needle cover straight off.
- Check the syringe for air bubbles. If there are air bubbles, gently tap the syringe with your fingers until the air bubbles rise to the top of the syringe. Slowly push the plunger up to force the air bubbles out of the syringe.
- Push the plunger up to the number (mL) on the syringe that matches the dose of NEUPOGEN that your doctor prescribed.
- Check again to make sure the correct dose of NEUPOGEN is in the syringe.
- Gently place the prefilled syringe with the window flat on your clean working surface so that the needle does not touch anything.
Selecting and preparing the injection site
- Choose an injection site. Four recommended injection sites for NEUPOGEN are:
- The outer area of your upper arms
- The abdomen, except for the two inch area around your navel
- The front of your middle thighs
- The upper outer areas of your buttocks
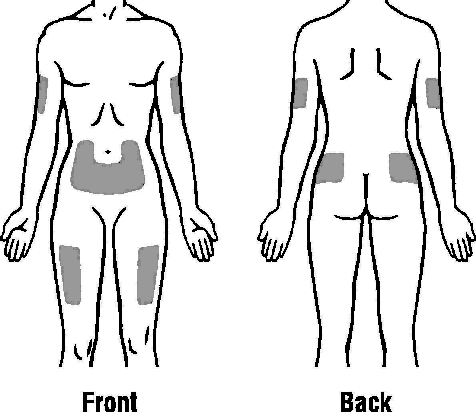
Choose a new site each time you inject NEUPOGEN . Choosing a new site can help avoid soreness at any one site. Do not inject NEUPOGEN into an area that is tender, red, bruised, or hard or that has scars or stretch marks.
- Clean the injection site with a new alcohol swab.
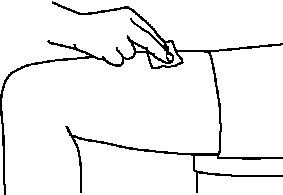
Injecting the dose of NEUPOGEN
- For injecting the dose of NEUPOGEN from a vial, remove the syringe and needle from the vial. For injecting the dose of NEUPOGEN from a prefilled syringe, pick up the prefilled syringe from your clean flat working surface by grabbing the sides of the needle guard with your thumb and forefinger.
- Hold the syringe in the hand you will use to inject NEUPOGEN . Use the other hand to pinch a fold of skin at the cleaned injection site. Note: If using a prefilled syringe with a needle guard, hold the syringe barrel through the needle guard windows when giving the injection.

- Holding the syringe like a pencil, use a quick dart-like motion to insert the needle either straight up and down (90 degree angle) or at a slight angle (45 degrees) into the skin.

- After the needle is inserted, let go of the skin. Pull the plunger back slightly. If no blood appears, slowly push down on the plunger all the way, until all the NEUPOGEN is injected. If blood comes into the syringe, do not inject NEUPOGEN , because the needle has entered a blood vessel. Withdraw the syringe and discard it in the puncture-proof container. Repeat the steps to prepare a new syringe (or get a new prefilled syringe) and choose and clean a new injection site. Remember to check again for blood before injecting NEUPOGEN .

- When the syringe is empty, pull the needle out of the skin and place a cotton ball or gauze over the injection site and press for several seconds.

- Use a prefilled syringe with the needle guard or a syringe and vial only once. If you are using a syringe, DO NOT put the needle cover (the cap) back on the needle. Discard the vial with any remaining NEUPOGEN liquid.
Activating the Needle Guard for the prefilled syringe after the injection has been given
- After injecting NEUPOGEN from the prefilled syringe, do not recap the needle. Keep your hands behind the needle at all times. While holding the clear plastic finger grip of the syringe with one hand, grasp the orange needle guard with your free hand and slide the orange needle guard over the needle until the needle is completely covered and the needle guard clicks into place. NOTE: If an audible click is not heard, the needle guard may not be completely activated.


- Place the prefilled syringe with the activated needle guard into a puncture-proof container for proper disposal as described below.
Disposal of syringes, needles, vials and needle guards
You should always follow the instructions given by your doctor, nurse, or pharmacist on how to properly dispose of containers with used syringes, needles, vials and needle guards. There may be special state and local laws for disposal of used needles and syringes.
- Place all used needles, needle covers, syringes, and vials (empty or unused contents) into a Sharps container given to you by your doctor or pharmacist or in a hard-plastic container with a screw-on cap, or a metal container with a plastic lid, such as a coffee can, labeled used syringes. If a metal container is used, cut a small hole in the plastic lid and tape the lid to the metal container. If a hard-plastic container is used, always screw the cap on tightly after each use.
- Do not use glass or clear plastic containers.
- When the container is full, tape around the cap or lid to make sure the cap or lid does not come off. Do not throw the container in the household trash. Do not recycle.
- Always keep the container out of the reach of children.
How should NEUPOGEN be stored?
NEUPOGEN should be stored in the refrigerator at 2 to 8 C (36 to 46 F), but not in the freezer. Avoid shaking NEUPOGEN . If NEUPOGEN is accidentally frozen, allow it to thaw in the refrigerator before giving the next dose. However, if it is frozen a second time, do not use it and contact your doctor or nurse for further instructions. NEUPOGEN can be left out at room temperature for up to 24 hours. Do not leave NEUPOGEN in direct sunlight. If you have any questions about storage or how to carry NEUPOGEN when you travel, contact your doctor, nurse, or pharmacist.
What are the ingredients in NEUPOGEN ?
Each syringe and vial contains Filgrastim in a sterile, clear, colorless, preservative-free solution containing acetate, sorbitol, polysorbate 80, and sodium.
The needle cover on the single-use prefilled syringe contains dry natural rubber (latex), which should not be handled by persons sensitive to this substance.
[Amgen Logo]
Manufactured by:
Amgen Manufacturing, Limited,
a subsidiary of Amgen Inc.
One Amgen Center Drive
Thousand Oaks, California 91320-1799 U.S.A.3xxxxxx
1991-2007 Amgen Inc. All rights reserved.
v8.1 - Issue Date: 09/2007
Preventative Healthcare Newsletter
Subscribe to receive exciting health news that can extend your lifespan and improve quality of life.


















