^
Principal Display Panel - 40 mg Tolerance Test Carton Label
Rx Only
NDC 10122-212-04
Bronchitol ®
(mannitol) inhalation powder
Tolerance Test
40 mg per capsule
FOR ORAL INHALATION ONLY
Contents:
The Bronchitol Tolerance Test MUST be completed toidentify bronchial hyperresponsiveness to mannitolprior to prescribing Bronchitol. Read Tolerance Testinstructions for detailed procedure.
Do Not Swallow Bronchitol Capsules
Chiesi
^ Pharmacodynamics
The pharmacodynamics of mannitol are unknown.
^ Use And Maintenance Of Inhaler
Instruct patients on safe hygiene practices (clean and dry hands thoroughly) and correct inhaler use, including loading of capsules and proper inhalation technique per the Patient Instructions for Use.
The BRONCHITOL inhaler should be discarded and replaced after 7 days of use. If the inhaler does need to be washed, the patient should allow the inhaler to thoroughly air dry before next use.
^ Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information and Patient Instructions for Use).
BRONCHITOL Tolerance Test
Inform patients that a BRONCHITOL Tolerance Test is required prior to beginning treatment with BRONCHITOL. The BRONCHITOL Tolerance Test must be performed by a healthcare practitioner equipped to monitor oxygen saturation (SpO2), perform spirometry (FEV1), and manage acute bronchospasm.
Inhaled Short-Acting Bronchodilator Use
Instruct patients that an inhaled short-acting bronchodilator such as albuterol must always be administered 5 to 15 minutes prior to every dose of BRONCHITOL.
Bronchospasm
Prior to administration, inform patients that bronchospasm can occur with inhalation of BRONCHITOL. If patient experiences bronchospasm, instruct the patient to discontinue BRONCHITOL and contact their healthcare practitioner right away.
Hemoptysis
Inform patients that hemoptysis can occur with inhalation of BRONCHITOL. If a patient experiences hemoptysis, instruct patients to discontinue BRONCHITOL and contact their healthcare practitioner right away.
Administration
Instruct patients on the proper administration of BRONCHITOL with the inhaler. The recommended dosage is 10 capsules (400 mg) twice a day. This requires inhaling the contents of 10 capsules administered individually once in the morning and once at least 2-3 hours before bed.
Manufactured by:
Pharmaxis Ltd
20 Rodborough Rd
Frenchs Forest NSW 2086 AUSTRALIA
Manufactured for:
Chiesi USA, Inc.
Cary, NC 27518
USA
BRONCHITOL® is a registered trademark of Pharmaxis Ltd
CTBR-001-1120-01-SPL-1
^ Description
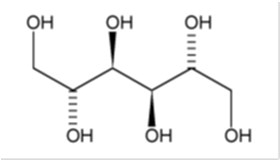
BRONCHITOL (mannitol) inhalation powder contains D-Mannitol (referred to throughout as mannitol) as the active ingredient. Mannitol is a hexahydric sugar alcohol, with the following chemical name hexane-1,2,3,4,5,6-hexol and chemical structure:
Mannitol is a white or almost white crystalline powder or free-flowing granules with an empirical formula of C6H14O6 and molecular weight of 182.2. Mannitol is freely soluble in water, and very slightly soluble in alcohol. Mannitol shows polymorphism.
BRONCHITOL contains mannitol powder spray dried into particles of respirable size filled into clear, colorless hard gelatin capsules. There are no inactive ingredients in BRONCHITOL.
The accompanying white plastic inhaler is comprised of a mouthpiece, blue piercing buttons, capsule chamber, and a removable cap. A blister pack consists of 10 capsules, each containing 40 mg mannitol. After a capsule is placed in the capsule chamber and pierced by firmly pressing and releasing the buttons on the side of the device, the powder within the capsule becomes exposed and ready for dispersion into the airstream generated by the patient upon inhalation through the mouthpiece. Under standardized in vitro test conditions, the inhaler delivers 32.2 mg of mannitol per inhalation when tested at a flow rate of 60 L/min for 2 seconds. The actual amount of drug delivered to the lungs will depend on patient factors, such as inspiratory flow profile.
^ Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The overall safety profile for BRONCHITOL is based on data from 1,020 CF patients from three 26-week, randomized, double-blind, controlled trials (Trials 1, 2, and 3). While CF patients aged 6 to 17 years were included in two of the three trials, BRONCHITOL is not indicated for use in this age group [see Indications (1), Use in Specific Populations (8)]. The safety data described below are based on 761 adult patients who received at least one dose of study drug in the three trials.
Of the 761 adult patients, 45% of patients were female and 98% were Caucasian; 414 received BRONCHITOL and 347 received control (50 mg inhaled mannitol) for up to 26 weeks. Adult patients treated with BRONCHITOL were ages 18 to 59 years with a mean baseline FEV1 of 62.0% of predicted.
In these three trials, the proportion of adult patients who prematurely discontinued study drug due to adverse reactions was 12.3% for patients treated with BRONCHITOL and 8.6% for patients treated with control. Serious adverse reactions occurred in 18.8% of patients treated with BRONCHITOL and 18.4% of patients treated with control. Serious adverse reactions occurring with greater than 1% incidence and more frequently in BRONCHITOL-treated adult patients compared to control-treated patients were CF exacerbations (13.3% vs. 11.2%), hemoptysis (1.4% vs. 1.2%) and lower respiratory tract infection (1.2% vs 0.9%).
The incidence of Adverse Reactions in adults during the 26 week treatment period with BRONCHITOL across the three trials is shown in Table 1.
In Trials 1, 2, and 3, exacerbations of cystic fibrosis (reported as condition aggravated) occurred in 132 of 414 (32%) adult patients receiving BRONCHITOL and in 114 of 347 (33%) adult patients receiving control (50 mg inhaled mannitol). Exacerbations of cystic fibrosis reported as serious adverse reactions occurred in 55 of 414 adult patients (13%) receiving BRONCHITOL and in 39 of 347 adult patients (11%) receiving control. Within the U.S. adult subgroup (comprising 27% of adults enrolled), exacerbations of cystic fibrosis reported as serious adverse reactions occurred in 23 of 110 (21%) patients receiving BRONCHITOL and in 10 of 93 (11%) patients receiving control. Among the non-U.S. adult subgroup (comprising 73% of adults enrolled), exacerbations of cystic fibrosis reported as serious adverse reactions occurred in 11% of patients in each treatment arm.
^ Recommended Dosage For Treatment Of Cystic Fibrosis
For patients who have passed the BTT, the recommended dosage of BRONCHITOL is 400 mg twice a day by oral inhalation (the contents of 10 capsules administered individually) via the inhaler [see Dosage and Administration (2.1)].
A short-acting bronchodilator should be administered by oral inhalation, 5-15 minutes before every dose of BRONCHITOL.
BRONCHITOL should be taken once in the morning and once in the evening, with the later dose taken at least 2-3 hours before bedtime.
^ Overdosage
Susceptible persons may experience bronchoconstriction from an overdosage. If excessive coughing and bronchoconstriction occurs, immediately administer an inhaled short-acting bronchodilator and other medical treatments as necessary.
^ Lactation
Risk Summary
It is not known whether BRONCHITOL is excreted in human breast milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for BRONCHITOL and any potential adverse effects on the breastfed child from BRONCHITOL or from the underlying maternal condition.
^3 Dosage Forms And Strengths
Inhalation powder: 40 mg mannitol per capsule; clear, colorless hard gelatin capsule imprinted with “PXS 40 mg”
^ Pediatric Use
BRONCHITOL is not indicated for use in children and adolescents. The safety and effectivenss of BRONCHITOL have not been established in pediatric patients for cystic fibrosis. Patients aged 6 years to 17 years were included in two 26-week, double-blind clinical trials (Trials 2 and 3). In these trials, 154 patients under 18 years of age received BRONCHITOL and 105 patients received control (50 mg inhaled mannitol). Hemoptysis was reported in 12 of 154 (7.8%) patients who received BRONCHITOL and in 2 of 105 (1.9%) patients who received control.
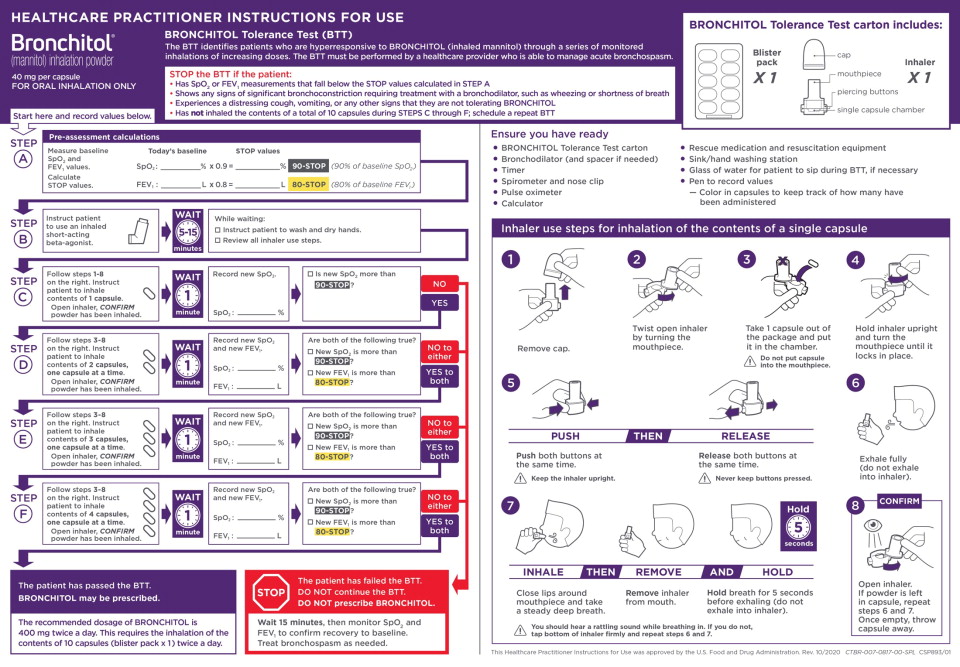
^ Required Testing and Evaluation Prior To Prescribing Bronchitol (bronchitol Tolerance Test)
Prior to prescribing BRONCHITOL for treatment of cystic fibrosis, the BRONCHITOL Tolerance Test (BTT) must be administered and performed under the supervision of a healthcare practitioner who is able to manage acute bronchospasm, to identify patients who are suitable candidates for BRONCHITOL maintenance therapy.
See the BTT Healthcare Practitioner (HCP) Instructions for Use (IFU) for complete instructions and to avoid medication errors associated with BTT dosing and procedures.
Do not use BRONCHITOL add-on maintenance therapy in patients who fail the BTT [see Contraindications (4)].
^4 Contraindications
BRONCHITOL is contraindicated in the following conditions:
^ How Supplied/storage And Handling
BRONCHITOL (mannitol) inhalation powder:
BRONCHITOL should be stored between 68°F-77°F (20°C-25°C) with excursions permitted between 59°F-86°F (15°C-30°C). [See USP Controlled Room Temperature]. Do not refrigerate. Do not freeze.
The Training Kit (NDC 10122-216-01), containing empty gelatin capsules, should be stored between 68°F-77°F (20°C-25°C) with excursions permitted from 59°F-86°F (15°C-30°C).
BRONCHITOL should only be used with the provided inhaler, which is a white plastic inhaler comprised of a mouthpiece, blue piercing buttons, capsule chamber, and a removable cap. All remaining unused (opened and unopened) blister packs and the inhalers should be properly discarded. Be sure to read the accompanying BRONCHITOL instructions completely before administration. If you have any questions, contact the supplier at 1-888-661-9260.
^ Geriatric Use
Clinical trials of BRONCHITOL did not include sufficient numbers of patients with cystic fibrosis who were 65 years of age and older to allow evaluation of safety and efficacy in this population.
^ Carcinogenesis, Mutagenesis, Impairment Of Fertility
Carcinogenesis
In 2-year carcinogenicity studies in rats and mice mannitol did not show evidence of carcinogenicity at oral dietary concentrations up to 5% (or 7,500 mg/kg on a mg/kg basis). These doses were approximately 55 and 30 times the MRHDID, respectively, on a mg/m2 basis.
Mutagenesis
Mannitol tested negative in the following assays: bacterial gene mutation assay, in vitro mouse lymphoma assay, in vitro chromosomal aberration assay in WI-38 human cells, in vivo chromosomal aberration assay in rat bone marrow, in vivo dominant lethal assay in rats, and in vivo mouse micronucleus assay.
Impairment of Fertility
The effect of inhaled mannitol on fertility has not been investigated.
^ Hepatic And Renal Impairment
Clinical trials of BRONCHITOL did not include patients with hepatic or renal impairment. No specific dose recommendations for these patient populations are available. However, an increase in systemic exposure of mannitol can be expected in patients with renal impairment based on the kidney being its primary route of elimination.
^ Bronchospasm
BRONCHITOL Tolerance Test
BRONCHITOL can cause bronchospasm, which can be severe in susceptible individuals. Because of the risk of bronchospasm, prior to prescribing BRONCHITOL, perform the BRONCHITOL Tolerance Test (BTT), to identify patients who are appropriate for maintenance treatment with BRONCHITOL. The BTT must be administered under the supervision of a healthcare practitioner who can treat severe bronchospasm. In clinical trials, 896 adult patients with cystic fibrosis underwent the BTT and 72 patients (8%) failed or did not complete the BTT. Do not prescribe BRONCHITOL if the patient fails the BTT.
Maintenance Therapy
Bronchospasm may occur during inhalation of BRONCHITOL, even in patients who have passed the BTT. An inhaled short-acting bronchodilator must be administered 5-15 minutes before administration of each dose during maintenance therapy. In clinical studies, bronchospasm or bronchial hyperreactivity was reported in 4 of 414 adult patients (1.0%) receiving BRONCHITOL as maintenance therapy and in 2 of 347 adult patients (0.6%) receiving control (50 mg inhaled mannitol), even though these patients had passed the BTT. If bronchospasm occurs following dosing of BRONCHITOL, it should immediately be discontinued and treated with an inhaled short-acting bronchodilator or as medically appropriate.
^ Clinical Studies
The efficacy of BRONCHITOL for the treatment of cystic fibrosis (CF) was evaluated in 3 randomized, double-blind, controlled trials (Trials 1, 2, and 3).
All three trials were 26-week, randomized, double-blind, controlled studies in patients with CF. Trial 1 (NCT02134353) evaluated patients 18 years of age or older with baseline FEV1>40% to <90% of predicted. Trial 2 (NCT00446680) evaluated patients 6 years of age or older with baseline FEV1 >30% to <90% of predicted. Trial 3 (NCT00630812) evaluated pateints 6 years of age or older with baseline FEV1 >40% to <90% of predicted. All three trials excluded CF patients with an episode of hemoptysis (>60 mL) in the 3 months prior to enrollment. The use of inhaled hypertonic saline was not permitted in any of the three trials, but continued use of their other standard of care CF therapies were allowed (e.g., bronchodilators, inhaled antibiotics, and dornase alfa). While CF patients aged 6 to 17 years were included in Trials 2 and 3, BRONCHITOL is not indicated for use in this age group [see Indications (1), Warnings and Precautions (5), Adverse Reactions (6), Use in Specific Populations (8)].
Patients were randomized to receive either BRONCHITOL 400 mg or control (50 mg inhaled mannitol) twice daily. Each dose of BRONCHITOL was preceded by use of an inhaled short-acting bronchodilator (albuterol or equivalent) taken 5 to 15 minutes prior to initiation of BRONCHITOL dosing. The primary efficacy endpoint in all three studies was improvement in lung function as determined by the mean change from baseline in pre-dose FEV1 (mL) over 26 weeks of treatment and was analyzed using the pattern mixture model with multiple imputation.
Trial 1 evaluated 423 adult patients with a mean age of 28 years and a mean FEV1 63.9% predicted (range: 40.3% = minimum, 89.6% = maximum).
Treatment with BRONCHITOL resulted in a statistically significant improvement in FEV1. In Trial 1, the treatment difference between BRONCHITOL and control for the adjusted mean change in FEV1 from baseline over 26 weeks was 51 mL (95% CI 6 to 97 mL) shown in Table 2.
Trials 2 and 3 evaluated 295 and 305 patients, respectively. For the adjusted mean difference in the change from baseline in FEV1 over 26 weeks in the intention-to-treat population in Trials 2 and 3, the treatment difference between BRONCHITOL and Control was 68 mL (95% CI: 24 to 113 mL) and 52 mL (95% CI: -3 to 107 mL), respectively.
Post-hoc descriptive analyses of the adult subgroups from Trials 2 and 3 were performed. The adult subgroup analyses in Trial 2 and 3 evaluated 209 and 157 adult patients, respectively. In Trial 2, there was an adjusted mean difference in the change from baseline in FEV1 over 26 weeks in the intention-to-treat population of adults of 78 mL (95% CI: 21 to 135 mL). In Trial 3, there was an adjusted mean difference in the change from baseline in FEV1 over 26 weeks in the intention-to-treat population of adults of 78 mL (95% CI: 2 to 153 mL).
^ Pregnancy
Risk Summary
There are no adequate and well-controlled studies of BRONCHITOL in pregnant women. The available data on BRONCHITOL use in pregnant women are not sufficient to inform any drug-associated risks for major birth defects and miscarriage. Based on animal reproduction studies, no evidence of structural alterations was observed when mannitol was administered to pregnant rats and mice during organogenesis at doses up to approximately 20 and 10 times, respectively, the maximum recommended daily inhalation dose (MRDID) in humans [see Data]. There are risks to the mother associated with cystic fibrosis in pregnancy [see Clinial Considerations]. BRONCHITOL should be used during pregnancy only if the potential benefit justifies the potential risk to the mother and fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the United States general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Cystic fibrosis may increase the risk for preterm delivery.
Data
Animal Data
In animal reproduction studies, oral administration of mannitol to pregnant rats and mice during the period of organogenesis did not cause fetal structural alterations. The mannitol dose in rats and mice was approximately 20 and 10 times the maximum recommended human daily inhalation dose (MRDID) in humans, respectively, (on a mg/m2 basis at maternal doses of 1600 mg/kg/day in both species).
^6 Adverse Reactions
The following clinically significant adverse reactions are described elsewhere in the labeling:
^7 Drug Interactions
No formal drug interaction studies have been conducted with mannitol, the active ingredient in BRONCHITOL.
^ hemoptysis
Hemoptysis may occur with BRONCHITOL use. Hemoptysis was reported in 43 (10.4%) adult patients receiving BRONCHITOL and in 33 (9.5%) adult patients receiving control (50 mg inhaled mannitol) during clinical studies. In patients aged 6 years to 17 years, hemoptysis was reported in 12 of 154 (7.8%) patients who received BRONCHITOL and in 2 of 105 (1.9%) patients who received control. BRONCHITOL has not been studied in patients with a history of episodes of significant hemoptysis (volume greater than 60 mL) in the previous 3 months. BRONCHITOL should be discontinued in the event of hemoptysis. BRONCHITOL is not indicated for use in children and adolescents.
^ Pharmacokinetics
Absorption
Following oral inhalation of 635 mg, the mean mannitol peak plasma concentration (Cmax) was 13.71 mcg/mL while the mean extent of systemic exposure (AUC) was 73.15 mcg•hr/mL. The mean time to peak plasma concentration (Tmax) after oral inhalation was 1.5 hour.
Distribution
Based on intravenous administration, the volume of distribution of mannitol was 34.3 L.
Elimination
Metabolism
Mannitol is metabolized in a CYP-independent manner through the glycolytic pathway via dehydrogenation to fructose. The extent of metabolism of mannitol appears to be small. This is evident from a urinary excretion of about 87% of unchanged drug after an intravenous dose to healthy patients.
Excretion
Following oral inhalation, the elimination half-life of mannitol was 4.7 hours. The mean terminal elimination half-life for mannitol in plasma remained unchanged regardless of the route of administration (oral, inhalation, and intravenous). The urinary excretion rate versus time profile for mannitol was consistent for all routes of administration. The total clearance after intravenous administration was 5.1 L/hr while the renal clearance was 4.4 L/hr. Therefore, the clearance of mannitol was predominately via the kidney. Following inhalation of 635 mg of mannitol in 18 healthy patients, about 55% of the total dose was excreted in the urine as unchanged mannitol. Following oral or intravenous administration of a 500 mg dose, the corresponding values were 54% and 87% of the dose, respectively.
Specific Populations
Patients with Hepatic and Renal Impairment: Formal pharmacokinetic studies using BRONCHITOL have not been conducted in patients with hepatic or renal impairment. Since the drug is eliminated primarily via the kidney, an increase in systemic exposure can be expected in renally impaired patients.
Drug Interaction Studies
No formal drug interaction studies have been conducted with BRONCHITOL.
^Healthcare Practitioner Instructions For Use
^1 Indications And Usage
BRONCHITOL is indicated as add-on maintenance therapy to improve pulmonary function in adult patients 18 years and older with Cystic Fibrosis. Use BRONCHITOL only for adults who have passed the BRONCHITOL Tolerance Test [see Dosage and Administration (2.1)].
^ Mechanism Of Action
The precise mechanism of action of BRONCHITOL in improving pulmonary function in cystic fibrosis patients is unknown.