^ Pharmacodynamics
Memantine showed low to negligible affinity for GABA, benzodiazepine, dopamine, adrenergic, histamine and glycine receptors and for voltage-dependent Ca , Na or K channels. Memantine also showed antagonistic effects at the 5HT receptor with a potency similar to that for the NMDA receptor and blocked nicotinic acetylcholine receptors with one-sixth to one-tenth the potency. 2+ + + 3
studies have shown that memantine does not affect the reversible inhibition of acetylcholinesterase by donepezil, galantamine, or tacrine. In vitro
^ Seizures
NAMENDA XR has not been systematically evaluated in patients with a seizure disorder. In clinical trials of memantine, seizures occurred in 0.3% of patients treated with memantine and 0.6% of patients treated with placebo.
^ Dosage Form
Capsule:
Each capsule contains 7 mg, 14 mg, 21 mg or 28 mg of memantine HCl.
The 7 mg capsules are a yellow opaque #4 size capsule, with “FLI 7 mg” black imprint.
The 14 mg capsules are a yellow cap and dark green opaque body #4 size capsule, with “FLI 14 mg” black imprint on the yellow cap.
The 21 mg capsules are a white to off-white cap and dark green opaque body #4 size capsule, with “FLI 21 mg” black imprint on the white to off-white cap.
The 28 mg capsules are a dark green opaque #3 size capsule, with “FLI 28 mg” white imprint.
^ Vital Sign Changes
NAMENDA XR and placebo groups were compared with respect to (1) mean change from baseline in vital signs (pulse, systolic blood pressure, diastolic blood pressure, and weight) and (2) the incidence of patients meeting criteria for potentially clinically significant changes from baseline in these variables. There were no clinically important changes in vital signs in patients treated with NAMENDA XR. A comparison of supine and standing vital sign measures for NAMENDA XR and placebo in Alzheimer's patients indicated that NAMENDA XR treatment is not associated with orthostatic changes.
^ Dosage Strengths
For a full list of excipients, see . Description ( ) 11
^ Hypersensitivity
NAMENDA XR is contraindicated in patients with known hypersensitivity to memantine hydrochloride or to any excipients used in the formulation See . [ Description ( )] 11
^ Patient Counseling Information
See FDA-approved patient labeling.
To assure safe and effective use of NAMENDA XR, the information and instructions provided in the patient information section should be discussed with patients and caregivers.
Patients and caregivers should be instructed to take NAMENDA XR only once per day, as prescribed.
Patients and caregivers should be instructed that NAMENDA XR capsules be swallowed whole. Alternatively, NAMENDA XR capsules may be opened and sprinkled on applesauce and the entire contents should be consumed. The capsules should not be divided, chewed or crushed.
Patients and caregivers should be advised that the product may cause headache, diarrhea and dizziness.
Manufactured for:Forest Pharmaceuticals, Inc. Subsidiary of Forest Laboratories, Inc. St. Louis, MO 63045
Manufactured by: Forest Laboratories Ireland Ltd
Licensed from Merz Pharmaceuticals GmbH
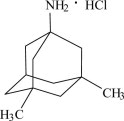
^ Description
NAMENDA XR is an orally active NMDA receptor antagonist. The chemical name for memantine hydrochloride is 1-amino-3,5-dimethyladamantane hydrochloride with the following structural formula:
The molecular formula is C H N•HCl and the molecular weight is 215.76. Memantine HCl occurs as a fine white to off-white powder and is soluble in water. 12 21
NAMENDA XR capsules are supplied for oral administration as 7, 14, 21 and 28 mg capsules (see Section ). Each capsule contains extended release beads with the labeled amount of memantine HCl and the following inactive ingredients: sugar spheres, polyvinylpyrrolidone, hypromellose, talc, polyethylene glycol, ethylcellulose, ammonium hydroxide, oleic acid, and medium chain triglycerides in hard gelatin capsules. How Supplied 16
^Renal Impairment
No dosage adjustment is recommended in patients with mild or moderate renal impairment.
A target dose of 14 mg/day is recommended in patients with severe renal impairment (creatinine clearance of 5 – 29 mL/min, based on the Cockroft-Gault equation).
^ Clinical Trial Data Sources
NAMENDA XR was evaluated in a double-blind placebo-controlled trial treating a total of 676 patients with moderate to severe dementia of the Alzheimer's type (341 patients treated with NAMENDA XR 28 mg/day dose and 335 patients treated with placebo) for a treatment period up to 24 weeks.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
^ Memantine Immediate Release Clinical Trial And Post Marketing Spontaneous Reports
The following additional adverse reactions have been identified from previous worldwide experience with memantine (immediate release) use. These adverse reactions have been chosen for inclusion because of a combination of seriousness, frequency of reporting, or potential causal connection to memantine and have not been listed elsewhere in labeling. However, because some of these adverse reactions were reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship between their occurrence and the administration of memantine. These events include:
agranulocytosis, leukopenia (including neutropenia), pancytopenia, thrombocytopenia. thrombotic thrombocytopenic purpura. Blood and Lymphatic System Disorders:
atrial fibrillation, atrioventricular block (including 2nd and 3rd degree block), cardiac failure, orthostatic hypotension, and torsades de pointes. Cardiac Disorders:
inappropriate antidiuretic hormone secretion. Endocrine Disorders:
colitis, pancreatitis. Gastrointestinal disorders:
malaise, sudden death. General disorders and administration site conditions:
hepatitis (including abnormal hepatic function test, cytolytic and cholestatic hepatitis), hepatic failure. Hepatobiliary Disorders:
sepsis. Infections and infestations:
electrocardiogram QT prolonged, international normalized ratio increased. Investigations:
hypoglycaemia, hyponatraemia. Metabolism and Nutrition Disorders:
convulsions (including grand mal), cerebrovascular accident, dyskinesia, extrapyramidal disorder, hypertonia, loss of consciousness, neuroleptic malignant syndrome, Parkinsonism, tardive dyskinesia, transient ischemic attack. Nervous System Disorders:
hallucinations (both visual and auditory), restlessness, suicidal ideation. Psychiatric Disorders:
acute renal failure (including abnormal renal function test), urinary retention. Renal and Urinary Disorders:
rash, Stevens Johnson syndrome. Skin Disorders:
pulmonary embolism, thrombophlebitis, deep venous thrombosis. Vascular Disorders:
The following adverse events have been reported to be temporally associated with memantine treatment and are not described elsewhere in the product labeling: aspiration pneumonia, bone fracture, carpal tunnel syndrome, cerebral infarction, chest pain, cholelithiasis, claudication, depressed level of consciousness (including rare reports of coma), dysphagia, encephalopathy, gastritis, gastroesophageal reflux, intracranial hemorrhage, hyperglycemia, hyperlipidemia, ileus, impotence, lethargy, myoclonus, supraventricular tachycardia, and tachycardia. However, there is again no evidence that any of these additional adverse events are caused by memantine.
^ Recommended Dosing
The dosage of NAMENDA XR shown to be effective in a controlled clinical trial is 28 mg once daily.
The recommended starting dose of NAMENDA XR is 7 mg once daily. The recommended target dose is 28 mg once daily. The dose should be increased in 7 mg increments to 28 mg once daily. The minimum recommended interval between dose increases is one week, and only if the previous dose has been well tolerated. The maximum recommended dose is 28 mg once daily.
NAMENDA XR can be taken with or without food. NAMENDA XR capsules can be taken intact or may be opened, sprinkled on applesauce, and thereby swallowed. The entire contents of each NAMENDA XR capsule should be consumed; the dose should not be divided.
Except when opened and sprinkled on applesauce, as described above, NAMENDA XR should be swallowed whole. NAMENDA XR capsules should not be divided, chewed, or crushed.
^ Overdosage
Signs and symptoms most often accompanying overdosage with other formulations of memantine in clinical trials and from worldwide marketing experience, alone or in combination with other drugs and/or alcohol, include agitation, asthenia, bradycardia, confusion, coma, dizziness, ECG changes, increased blood pressure, lethargy, loss of consciousness, psychosis, restlessness, slowed movement, somnolence, stupor, unsteady gait, visual hallucinations, vertigo, vomiting, and weakness. The largest known ingestion of memantine worldwide was 2 grams in an individual who took memantine in conjunction with unspecified antidiabetic medications. This person experienced coma, diplopia, and agitation, but subsequently recovered.
One patient participating in a NAMENDA XR clinical trial unintentionally took 112 mg of NAMENDA XR daily for 31 days and experienced an elevated serum uric acid, elevated serum alkaline phosphatase, and low platelet count.
No fatalities have been noted with overdoses of memantine alone. A fatal outcome has very rarely been reported when memantine has been ingested as part of overdosing with multiple drugs; in those instances, the relationship between memantine and a fatal outcome has been unclear.
Because strategies for the management of overdose are continually evolving, it is advisable to contact a poison control center to determine the latest recommendations for the management of an overdose of any drug. As in any cases of overdose, general supportive measures should be utilized, and treatment should be symptomatic. Elimination of memantine can be enhanced by acidification of urine.
^ Drugs That Make The Urine Alkaline
The clearance of memantine was reduced by about 80% under alkaline urine conditions at pH 8. Therefore, alterations of urine pH towards the alkaline condition may lead to an accumulation of the drug with a possible increase in adverse effects. Urine pH is altered by diet, drugs (e.g. carbonic anhydrase inhibitors, sodium bicarbonate) and clinical state of the patient (e.g. renal tubular acidosis or severe infections of the urinary tract). Hence, memantine should be used with caution under these conditions.
^ Use With Cholinesterase Inhibitors
Coadministration of memantine with the AChE inhibitor donepezil HCl did not affect the pharmacokinetics of either compound. In a 24-week controlled clinical study in patients with moderate to severe Alzheimer's disease, the adverse event profile observed with a combination of memantine immediate-release and donepezil was similar to that of donepezil alone.
^ Nursing Mothers
It is not known whether memantine is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when memantine is administered to a nursing mother.
^ Use With Other N-methyl-d-aspartate (nmda) Antagonists
The combined use of NAMENDA XR with other NMDA antagonists (amantadine, ketamine, and dextromethorphan) has not been systematically evaluated and such use should be approached with caution.
^ Pediatric Use
The safety and effectiveness of memantine in pediatric patients have not been established.
Memantine failed to demonstrate efficacy in two 12-week controlled clinical studies of 578 pediatric patients aged 6-12 years with autism spectrum disorders (ASD), including autism, Asperger's disorder, and Pervasive Development Disorder - Not Otherwise Specified (PDD-NOS). Memantine has not been studied in pediatric patients under 6 years of age or over 12 years of age. Memantine treatment was initiated at 3 mg/day and the dose was escalated to the target dose (weight-based) by week 6. Oral doses of memantine 3, 6, 9, or 15 mg extended-release capsules were administered once daily to patients with weights < 20 kg, 20-39 kg, 40-59 kg and ≥ 60 kg, respectively.
In a randomized, 12-week double-blind, placebo-controlled parallel study (Study A) in patients with autism, there was no statistically significant difference in the Social Responsiveness Scale (SRS) total raw score between patients randomized to memantine (n=54) and those randomized to placebo (n=53). In a 12-week responder-enriched randomized withdrawal study (Study B) in 471 patients with ASD, there was no statistically significant difference in the loss of therapeutic response rates between patients randomized to remain on full-dose memantine (n=153) and those randomized to switch to placebo (n=158).
The overall safety profile of memantine in pediatric patients was generally consistent with the known safety profile in adults [ ]. see Adverse Reactions (6.1)
In Study A, the treatment emergent adverse events in the memantine group (n=56) that were reported in at least 5% of patients and twice that in the placebo group (N=58) are listed in : Table 2
The treatment emergent adverse events that were reported in at least 5% of patients in the 12-48 week open-label study to identify responders to enroll in Study B are listed in : Table 3
In the randomized withdrawal study (Study B), the treatment emergent adverse event in patients randomized to placebo (n=160) and reported in at least 5% of patients and twice that of the full-dose memantine treatment group (n=157) was irritability (5.0% vs 2.5%).
In a juvenile animal study, male and female juvenile rats were administered memantine (15, 30, and 45 mg/kg/day) starting on postnatal day (PND) 14 through PND 70. Body weights were reduced at 45 mg/kg/day. Delays in sexual maturation were noted in male and female rats at doses ≥ 30 mg/kg/day. Memantine induced neuronal lesions in several areas of the brain on PND 15 and 17 at doses ≥ 30 mg/kg/day. Behavioral toxicity (decrease percent of auditory startle habituation) was noted for animals in the 45 mg/kg/day dose group. The 15 mg/kg/day dose was considered the No-Observed-Adverse-Effect-Level (NOAEL) for this study.
In a second juvenile rat toxicity study, male and female juvenile rats were administered memantine (1, 3, 8, 15, 30, and 45 mg/kg/day) starting on postnatal day (PND) 7 through PND 70. Due to early memantine-related mortality, the 30 and 45 mg/kg/day dose groups were terminated without further evaluation. Memantine induced apoptosis or neuronal degeneration in several areas of the brain on PND 8, 10, and 17 at a dose of 15 mg/kg/day. The NOAEL for apoptosis and neuronal degeneration was 8 mg/kg/day. Behavioral toxicity (effects on motor activity, auditory startle habituation, and learning and memory) was noted at doses ≥ 3 mg/kg/day during treatment, but was not seen after drug discontinuation. Therefore, the 1 mg/kg/day dose was considered the NOAEL for the neurobehavioral effect in this study.
^ How Supplied/storage And Handling
NDC:68151-5817-8 in a CUP of 1 CAPSULE, EXTENDED RELEASES
^9 Drug Abuse And Dependence
Memantine is not a controlled substance. Memantine is a low to moderate affinity uncompetitive NMDA antagonist that did not produce any evidence of drug-seeking behavior or withdrawal symptoms upon discontinuation in 3,254 patients who participated in clinical trials at therapeutic doses. Post marketing data, outside the U.S., retrospectively collected, has provided no evidence of drug abuse or dependence.
^ Adverse Reactions Leading To Discontinuation
In the placebo-controlled clinical trial of NAMENDA XR [See )], which treated a total of 676 patients, the proportion of patients in the NAMENDA XR 28 mg/day dose and placebo groups who discontinued treatment due to adverse events were 10.0% and 6.3%, respectively. The most common adverse reaction in the NAMENDA XR treated group that led to treatment discontinuation in this study was dizziness at a rate of 1.5%. Clinical Studies ( 14
^ Carcinogenesis, Mutagenesis, Impairment Of Fertility
There was no evidence of carcinogenicity in a 113-week oral study in mice at doses up to 40 mg/kg/day (7 times the maximum recommended human dose [MRHD] on a mg/m basis). There was also no evidence of carcinogenicity in rats orally dosed at up to 40 mg/kg/day for 71 weeks followed by 20 mg/kg/day (14 and 7 times the MRHD on a mg/m basis, respectively) through 128 weeks. 2 2
Memantine produced no evidence of genotoxic potential when evaluated in the or reverse mutation assay, an chromosomal aberration test in human lymphocytes, an cytogenetics assay for chromosome damage in rats, and the mouse micronucleus assay. The results were equivocal in an gene mutation assay using Chinese hamster V79 cells. in vitro S. typhimurium E. coli in vitro in vivo in vivo in vitro
No impairment of fertility or reproductive performance was seen in rats administered up to 18 mg/kg/day (6 times the MRHD on a mg/m basis) orally from 14 days prior to mating through gestation and lactation in females, or for 60 days prior to mating in males. 2
^ Animal Toxicology
Memantine induced neuronal lesions (vacuolation and necrosis) in the multipolar and pyramidal cells in cortical layers III and IV of the posterior cingulate and retrosplenial neocortices in rats, similar to those which are known to occur in rodents administered other NMDA receptor antagonists. Lesions were seen after a single dose of memantine. In a study in which rats were given daily oral doses of memantine for 14 days, the no-effect dose for neuronal necrosis was 4 times the maximum recommended human dose (MRHD of 28 mg/day) on a mg/m basis. 2
In a neurotoxicity study, female rats were given oral doses of memantine (3, 10, 30, 60 mg/kg/day) alone or in combination with donepezil (3, 10 mg/kg/day) for 28 days. When administered alone, memantine induced neurodegeneration only at 60 mg/kg/day; however, when administered in combination with 10 mg/kg/day donepezil, memantine induced neurodegeneration at doses of 30 and 60 mg/kg/day. When 60 mg/kg/day memantine and 10 mg/kg/day donepezil were administered in combination, the incidence and severity of neurodegeneration was increased compared to that with 60 mg/kg/day memantine alone or with 30 mg/kg/day memantine in combination with 10 mg/kg/day donepezil. In addition, the combination of 60 mg/kg/day memantine and 10 mg/kg/day donepezil was associated with widespread neurodegeneration in cortical areas (perirhinal, temporal, entorhinal, frontal, insular, piriform) and in olfactory nucleus and subiculum, whereas in the other affected groups, there was limited cortical (entorhinal, retrosplenial) involvement. At the no-effect level of the combination (10 mg/kg/day memantine + 10 mg/kg/day donepezil), plasma exposures of memantine were similar to (AUC) or two times (Cmax) those expected in humans at the MRHD; plasma exposures of donepezil were 3 (AUC) or 6 (Cmax) times those in humans at the MRHD of donepezil (10 mg/day). In a published study, similar donepezil-mediated exacerbation of memantine-induced neurodegeneration was observed in female rats given single doses of memantine in combination with donepezil, both administered by intraperitoneal injection.
The potential for induction of central neurodegenerative lesions by NMDA receptor antagonists in humans is unknown.
^Namenda Xr (memantine Hydrochloride) Capsule, Extended Release Namenda Xr (memantine Hydrochloride) Kit

^ Effect Of Memantine On The Metabolism Of Other Drugs
studies conducted with marker substrates of CYP450 enzymes (CYP1A2, -2A6, -2C9, -2D6, -2E1, -3A4) showed minimal inhibition of these enzymes by memantine. In addition, i studies indicate that at concentrations exceeding those associated with efficacy, memantine does not induce the cytochrome P450 isozymes CYP1A2, -2C9, -2E1 and -3A4/5. No pharmacokinetic interactions with drugs metabolized by these enzymes are expected. In vitro n vitro
Pharmacokinetic studies evaluated the potential of memantine for interaction with donepezil (See ) and bupropion. Coadministration of memantine with the AChE inhibitor donepezil HCl does not affect the pharmacokinetics of either compound. Memantine did not affect the pharmacokinetics of the CYP2B6 substrate bupropion or its metabolite hydroxybupropion. Section Use with Cholinesterase Inhibitors 7.7
^ Drugs Highly Bound To Plasma Proteins
Because the plasma protein binding of memantine is low (45%), an interaction with drugs that are highly bound to plasma proteins, such as warfarin and digoxin, is unlikely [see Section .] 7
^ Clinical Studies
The effectiveness of NAMENDA XR as a treatment for patients with moderate to severe Alzheimer's disease was based on the results of a double-blind, placebo-controlled trial.
^ Effect Of Other Drugs On Memantine
Memantine is predominantly renally eliminated, and drugs that are substrates and/or inhibitors of the CYP450 system are not expected to alter the pharmacokinetics of memantine. A clinical drug-drug interaction study indicated that bupropion did not affect the pharmacokinetics of memantine.
^7 Drug Interactions
No drug-drug interaction studies have been conducted with NAMENDA XR, specifically.
^ Laboratory Changes
NAMENDA XR and placebo groups were compared with respect to (1) mean change from baseline in various serum chemistry, hematology, and urinalysis variables and (2) the incidence of patients meeting criteria for potentially clinically significant changes from baseline in these variables. These analyses revealed no clinically important changes in laboratory test parameters associated with NAMENDA XR treatment.
^ Drugs Eliminated Via Renal Mechanisms
Because memantine is eliminated in part by tubular secretion, coadministration of drugs that use the same renal cationic system, including hydrochlorothiazide (HCTZ), triamterene (TA), metformin, cimetidine, ranitidine, quinidine, and nicotine, could potentially result in altered plasma levels of both agents. However, coadministration of memantine and HCTZ/TA did not affect the bioavailability of either memantine or TA, and the bioavailability of HCTZ decreased by 20%. In addition, coadministration of memantine with the antihyperglycemic drug Glucovance (glyburide and metformin HCl) did not affect the pharmacokinetics of memantine, metformin and glyburide. Furthermore, memantine did not modify the serum glucose lowering effect of Glucovance , indicating the absence of a pharmacodynamic interaction. ® ®
^ Pharmacokinetics
Memantine is well absorbed after oral administration and has linear pharmacokinetics over the therapeutic dose range. It is excreted predominantly unchanged in urine and has a terminal elimination half-life of about 60-80 hours. In a study comparing 28 mg once daily NAMENDA XR to 10 mg twice daily NAMENDA C and AUC values were 48% and 33% higher for the XR dosage regimen, respectively. max 0-24
^1 Indications And Usage
NAMENDA XR (memantine hydrochloride) extended-release capsules are indicated for the treatment of moderate to severe dementia of the Alzheimer's type.
^ Genitourinary Conditions
Conditions that raise urine pH may decrease the urinary elimination of memantine resulting in increased plasma levels of memantine.
^ Most Common Adverse Reactions
The most commonly observed adverse reactions seen in patients administered NAMENDA XR in the controlled clinical trial, defined as those occurring at a frequency of at least 5% in the NAMENDA XR group and at a higher frequency than placebo were headache, diarrhea and dizziness.
lists treatment-emergent adverse reactions that were observed at an incidence of ≥ 2% in the NAMENDA XR treated group and occurred at a rate greater than placebo. Table 1
^ Other Adverse Reactions Observed During Clinical Trials Of Namenda Xr
Following is a list of treatment-emergent adverse reactions reported from 750 patients treated with NAMENDA XR for periods up to 52 weeks in double-blind or open-label clinical trials. The listing does not include those events already listed in , those events for which a drug cause was remote, those events for which descriptive terms were so lacking in specificity as to be uninformative, and those events reported only once which did not have a substantial probability of being immediately life threatening. Events are categorized by body system. Table 1
anemia. Blood and Lymphatic System Disorders:
bradycardia, myocardial infarction. Cardiac Disorders:
fecal incontinence, nausea. Gastrointestinal Disorders:
asthenia, fatigue, gait disturbance, irritability, peripheral edema, pyrexia. General Disorders:
bronchitis, nasopharyngitis, pneumonia, upper respiratory tract infection, urinary tract infection. Infections and Infestations:
fall. Injury, Poisoning and Procedural Complications:
weight decreased. Investigations:
anorexia, dehydration, decreased appetite, hyperglycemia. Metabolism and Nutrition Disorders:
arthralgia, pain in extremity. Musculoskeletal and Connective Tissue Disorders:
convulsion, dementia Alzheimer's type, syncope, tremor. Nervous System Disorders:
agitation, confusional state, delirium, delusion, disorientation, hallucination, insomnia, restlessness. Psychiatric Disorders:
cough, dyspnea. Respiratory, Thoracic and Mediastinal Disorders:
^ Ecg Changes
NAMENDA XR and placebo groups were compared with respect to (1) mean change from baseline in various ECG parameters and (2) the incidence of patients meeting criteria for potentially clinically significant changes from baseline in these variables. These analyses revealed no clinically important changes in ECG parameters associated with NAMENDA XR treatment.
^ Mechanism Of Action
Persistent activation of central nervous system N-methyl-D-aspartate (NMDA) receptors by the excitatory amino acid glutamate has been hypothesized to contribute to the symptomatology of Alzheimer's disease. Memantine is postulated to exert its therapeutic effect through its action as a low to moderate affinity uncompetitive (open-channel) NMDA receptor antagonist which binds preferentially to the NMDA receptor-operated cation channels. There is no evidence that memantine prevents or slows neurodegeneration in patients with Alzheimer's disease.